Get your patient on Tafluprost - Tafluprost Opthalmic solution/ Drops (Tafluprost Opthalmic)
Tafluprost - Tafluprost Opthalmic solution/ Drops prescribing information
INDICATIONS AND USAGE
Tafluprost ophthalmic solution is indicated for reducing elevated intraocular pressure in patients with open-angle glaucoma or ocular hypertension.
DOSAGE AND ADMINISTRATION
Apply one drop in the affected eye(s) once daily in the evening. (2 )
Recommended Dosage
The recommended dosage is one drop in the conjunctival sac of the affected eye(s) once daily in the evening.
Administration Instructions
Tafluprost ophthalmic solution should not be administered more than once daily since it has been shown that more frequent administration of prostaglandin analogs may lessen the intraocular pressure (IOP) lowering effect.
Reduction of the intraocular pressure starts approximately 2 to 4 hours after the first administration with the maximum effect reached after 12 hours.
Tafluprost ophthalmic solution may be used concomitantly with other topical ophthalmic drug products to lower IOP. If more than one topical ophthalmic product is being used, each one should be administered at least 5 minutes apart.
The solution from one single-dose container is to be used immediately after opening for administration to one or both eyes. Since sterility cannot be maintained after the single-dose container is opened, discard the open single-dose container and the remaining contents immediately after administration.
DOSAGE FORMS AND STRENGTHS
Ophthalmic solution: 0.0015% tafluprost in a single-dose container.
USE IN SPECIFIC POPULATIONS
Use in pediatric patients is not recommended because of potential safety concerns related to increased pigmentation following long-term chronic use. (8.4 )
Pregnancy
Risk Summary
There are no adequate and well-controlled studies of tafluprost administration in pregnant women to inform of drug-associated risks. In animal reproduction studies, intravenous administration of tafluprost to pregnant rabbits and rats throughout organogenesis resulted in embryofetal toxicities at exposures ≥5-times the human dose in rabbit and ≥2362-times the human dose in rat. (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population(s) is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In embryo-fetal development studies, intravenous administration of tafluprost to pregnant rats and rabbits during organogenesis caused increases in post-implantation losses in both species and reductions in fetal body weights in rats (≥0.03 mcg/kg/day in rabbits, 5-times the maximum clinical exposure based on C max ; ≥10 mcg/kg/day in rats, 2362-times the maximum clinical exposure based on C max ). Tafluprost also increased the incidence of vertebral skeletal abnormalities in rats and the incidence of skull, brain and spine malformations in rabbits at these same doses. In rats, there were no adverse effects on embryo-fetal development at a dose of 3 mcg/kg/day corresponding to maternal plasma levels of tafluprost acid that were 343 times the maximum clinical exposure based on C max . At the no-effect dose in rabbits (0.01 mcg/kg/day), maternal plasma levels of tafluprost acid were below the lower level of quantification (20 pg/mL).
In a pre- and postnatal development study, intravenous administration of tafluprost to pregnant rats during organogenesis and through birth and lactation, caused increased mortality of newborns, decreased body weights and delayed pinna unfolding in offsprings. The no observed adverse effect level was at a tafluprost intravenous dose of 0.3 mcg/kg/day which is greater than 3 times the maximum recommended clinical dose based on body surface area comparison.
Lactation
Risk Summary
There are no data on the presence of tafluprost or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production following topical ocular administration. Tafluprost and/or its metabolites are present in rat milk following ocular administration. When a drug is present in animal milk, it is likely that the drug will be present in human milk.
Pediatric Use
Use in pediatric patients is not recommended because of potential safety concerns related to increased pigmentation following long-term chronic use.
Geriatric Use
No overall clinical differences in safety or effectiveness have been observed between elderly and other adult patients.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Pigmentation
Tafluprost ophthalmic solution has been reported to cause changes to pigmented tissues. The most frequently reported changes have been increased pigmentation of the iris, periorbital tissue (eyelid) and eyelashes. Pigmentation is expected to increase as long as tafluprost is administered. The pigmentation change is due to increased melanin content in the melanocytes rather than to an increase in the number of melanocytes. After discontinuation of tafluprost, pigmentation of the iris is likely to be permanent, while pigmentation of the periorbital tissue and eyelash changes have been reported to be reversible in some patients. Patients who receive treatment should be informed of the possibility of increased pigmentation. The long term effects of increased pigmentation are not known.
Iris color change may not be noticeable for several months to years. Typically, the brown pigmentation around the pupil spreads concentrically towards the periphery of the iris and the entire iris or parts of the iris become more brownish. Neither nevi nor freckles of the iris appear to be affected by treatment. While treatment with tafluprost ophthalmic solution can be continued in patients who develop noticeably increased iris pigmentation, these patients should be examined regularly.
Eyelash Changes
Tafluprost may gradually change eyelashes and vellus hair in the treated eye. These changes include increased length, color, thickness, shape and number of lashes. Eyelash changes are usually reversible upon discontinuation of treatment.
Intraocular Inflammation
Tafluprost should be used with caution in patients with active intraocular inflammation (e.g., iritis/uveitis) because the inflammation may be exacerbated.
Macular Edema
Macular edema, including cystoid macular edema, has been reported during treatment with prostaglandin F2α analogs. Tafluprost should be used with caution in aphakic patients, in pseudophakic patients with a torn posterior lens capsule, or in patients with known risk factors for macular edema.
Contact Lens Use
Contact lenses should be removed prior to the administration of tafluprost ophthalmic solution and may be reinserted 15 minutes after administration.
ADVERSE REACTIONS
Most common ocular adverse reaction is conjunctival hyperemia (4% to 20%). (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Micro Labs USA Inc., at 1-855-839-8195 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Preservative-containing or nonpreserved tafluprost 0.0015% was evaluated in 905 patients in five controlled clinical studies of up to 24-months duration. The most common adverse reaction observed in patients treated with tafluprost was conjunctival hyperemia which was reported in a range of 4% to 20% of patients. Approximately 1% of patients discontinued therapy due to ocular adverse reactions.
Ocular adverse reactions reported at an incidence of ≥ 2% in these clinical studies included ocular stinging/irritation (7%), ocular pruritus including allergic conjunctivitis (5%), cataract (3%), dry eye (3%), ocular pain (3%), eyelash darkening (2%), growth of eyelashes (2%) and vision blurred (2%).
Nonocular adverse reactions reported at an incidence of 2% to 6% in these clinical studies in patients treated with tafluprost 0.0015% were headache (6%), common cold (4%), cough (3%) and urinary tract infection (2%).
Postmarketing Experience
The following adverse reactions have been identified during postapproval use of tafluprost. Because postapproval adverse reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Respiratory disorders: exacerbation of asthma, dyspnea
Eye disorders: iritis/uveitis
In postmarketing use with prostaglandin analogs, periorbital and lid changes including deepening of the eyelid sulcus have been observed.
DESCRIPTION
Tafluprost ophthalmic solution 0.0015% contains tafluprost, a fluorinated analog of prostaglandin F2α, for topical ophthalmic use. The chemical name for tafluprost is 1-methylethyl (5 Z )-7-{(1 R ,2 R ,3 R ,5 S )-2-[(1 E )-3,3-difluoro-4-phenoxy-1-butenyl}-3,5-dihydroxycyclopentyl]-5-heptenoate.
The molecular formula of tafluprost is C 25 H 34 F 2 O 5 and its molecular weight is 452.53.
Its structural formula is:
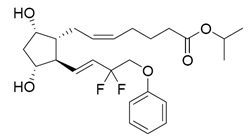
Tafluprost is a colorless to light yellow viscous liquid that is practically insoluble in water.
Tafluprost ophthalmic solution is supplied as a sterile solution with a pH range of 5.5 to 6.7 and an Osmolality range of 260 to 300 mOsmol/kg.
Tafluprost ophthalmic solution contains Active: tafluprost 0.015 mg/mL; Inactives: edetate disodium, glycerin, monobasic sodium phosphate dihydrate, polysorbate 80, hydrochloric acid and sodium hydroxide (to adjust pH) and water for injection.
Tafluprost ophthalmic solution does not contain a preservative.
CLINICAL PHARMACOLOGY
Mechanism of Action
Tafluprost acid, a prostaglandin analog, is a selective FP prostanoid receptor agonist which is believed to reduce intraocular pressure by increasing uveoscleral outflow. The exact mechanism of action is unknown at this time.
Pharmacokinetics
Absorption
Following instillation, tafluprost is absorbed through the cornea and is hydrolyzed to the biologically active acid metabolite, tafluprost acid. Following instillation of one drop of the 0.0015% solution once daily into each eye of healthy volunteers, the plasma concentrations of tafluprost acid peaked at a median time of 10 minutes on both Days 1 and 8. The mean plasma C max of tafluprost acid were 26 pg/mL and 27 pg/mL on Day 1, and Day 8, respectively. The mean plasma AUC estimates of tafluprost acid were 394 pg • min/mL and 432 pg • min/mL on day 1 and 8, respectively.
Elimination
Metabolism
Tafluprost, an ester prodrug, is hydrolyzed to its biologically active acid metabolite in the eye. The acid metabolite is further metabolized via fatty acid β-oxidation and phase II conjugation.
Mean plasma tafluprost acid concentrations were below the limit of quantification of the bioanalytical assay (10 pg/mL) at 30 minutes following topical ocular administration of tafluprost 0.0015% ophthalmic solution.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Tafluprost was not carcinogenic when administered subcutaneously daily for 24 months at doses up to 30 mcg/kg/day in rats and for 18 months at doses up to 100 mcg/kg/day in mice (over 1600 and 1300 times, respectively, the maximum clinical exposure based on plasma AUC).
Mutagenesis
Tafluprost was not mutagenic or clastogenic in a battery of genetic toxicology studies, including an in vitro microbial mutagenesis assay, an in vitro chromosomal aberration assay in Chinese hamster lung cells, and an in vivo mouse micronucleus assay in bone marrow.
Impairment of Fertility
In rats, no adverse effects on mating performance or fertility were observed with intravenous dosing of tafluprost at a dose of 100 mcg/kg/day (over 14000 times the maximum clinical exposure based on plasma C max or over 3600 times based on plasma AUC).
CLINICAL STUDIES
In clinical studies up to 24 months in duration, patients with open-angle glaucoma or ocular hypertension and baseline pressure of 23 to 26 mmHg who were treated with tafluprost dosed once daily in the evening demonstrated reductions in intraocular pressure at 3 and 6 months of 6 to 8 mmHg and 5 to 8 mmHg, respectively.
HOW SUPPLIED/STORAGE AND HANDLING
Tafluprost ophthalmic solution 0.0015% is supplied as 0.3 mL a sterile solution in translucent low density polyethylene single-dose containers packaged in foil pouches (15 single-dose containers per pouch). Each single-dose container has 0.3 mL solution corresponding to 0.0045 mg tafluprost.
Unit-of-Use Carton of 15 NDC 42571-264-26
Unit-of-Use Carton of 30 NDC 42571-264-73
Unit-of-Use Carton of 90 NDC 42571-264-74
Storage:
Store refrigerated at 2° to 8°C (36° to 46°F). During shipment tafluprost ophthalmic solution may be maintained at temperatures up to 40°C (104°F) for a period not exceeding 2 days. Mail-order prescriptions received after two days of the dispensing date noted in the prescribing label should not be used. Store in the original pouch. After the pouch is opened, unopened single-dose containers may be stored in the opened foil pouch for up to 30 days at room temperature 20° to 25°C (68° to 77°F). Protect from moisture. Write down the date you open the foil pouch in the space provided on the pouch. Discard any unopened containers 30 days after first opening the pouch.
Mechanism of Action
Tafluprost acid, a prostaglandin analog, is a selective FP prostanoid receptor agonist which is believed to reduce intraocular pressure by increasing uveoscleral outflow. The exact mechanism of action is unknown at this time.