Get your patient on Rytelo (Imetelstat Sodium)
Rytelo patient education
Patient toolkit
Dosage & administration

Rytelo prescribing information
INDICATIONS AND USAGE
RYTELO is indicated for the treatment of adult patients with low- to intermediate-1 risk myelodysplastic syndromes (MDS) with transfusion-dependent anemia requiring 4 or more red blood cell units over 8 weeks who have not responded to or have lost response to or are ineligible for erythropoiesis-stimulating agents (ESA).
DOSAGE AND ADMINISTRATION
Recommended Dosage
The recommended dosage of RYTELO is 7.1 mg/kg administered as an intravenous infusion over 2 hours every 4 weeks. Discontinue RYTELO if a patient does not experience a decrease in red blood cell (RBC) transfusion burden after 24 weeks of treatment (administration of 6 doses) or if unacceptable toxicity occurs at any time [see Dosage and Administration (2.3) ] .
Recommended Premedications
Administer the following pre-treatment medications at least 30 minutes prior to dosing to prevent or reduce potential infusion-related reactions:
- diphenhydramine (or equivalent) 25 mg to 50 mg, intravenously or orally
- hydrocortisone (or equivalent) 100 mg to 200 mg, intravenously or orally
Monitor patients for adverse reactions for at least one hour after the infusion has been completed [see Warnings and Precautions (5.3) and Adverse Reactions (6.1) ] .
Dosage Modifications for Adverse Reactions
Recommended dose reductions for Grade 3 and Grade 4 adverse reactions are found in Table 1.
The management of Grade 3 and Grade 4 adverse reactions may require temporary dose delay, dose reduction, or treatment discontinuation and are presented in Table 2 and Table 3. RYTELO treatment should be permanently discontinued if the patient cannot tolerate the lowest dose level of 4.4 mg/kg.
| Dose Reduction | Dose Every 4 Weeks |
|---|---|
| First dose reduction | 5.6 mg/kg |
| Second dose reduction | 4.4 mg/kg |
Dosage Modifications for Hematologic (Grade 3 and Grade 4) Adverse Reactions
Monitor complete blood cell counts prior to administration of RYTELO, weekly for the first two cycles, prior to each cycle thereafter, and as clinically indicated. Delay the next cycle if absolute neutrophil count is less than 1 × 10 9 /L or platelets are less than 50 × 10 9 /L. Modify dose as described in Table 2.
| Adverse Reaction | Severity Grade Severity based on National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03. | Occurrence | Treatment Modification |
|---|---|---|---|
| Abbreviation: ANC = absolute neutrophil count | |||
| Thrombocytopenia [see Warnings and Precautions (5.1) ] | Grade 3 | First | Delay RYTELO until recovery of platelets to 50 × 10 9 /L; restart at same dose level. |
| Second and Third | Delay RYTELO until recovery of platelets to 50 × 10 9 /L; restart at one dose level lower. | ||
| Fourth | Discontinue RYTELO. | ||
| Grade 4 | First and Second | Delay RYTELO until recovery of platelets to 50 × 10 9 /L; restart at one dose level lower. | |
| Third | Discontinue RYTELO. | ||
| Neutropenia [see Warnings and Precautions (5.2) ] | Grade 3 | First | Delay RYTELO until recovery of ANC to 1 × 10 9 /L; restart at same dose level. |
| Second and Third | Delay RYTELO until recovery of ANC to 1 × 10 9 /L; restart at one dose level lower. | ||
| Fourth | Discontinue RYTELO. | ||
| Grade 4 | First and Second | Delay RYTELO until recovery of ANC to 1 × 10 9 /L; restart at one dose level lower. | |
| Third | Discontinue RYTELO. | ||
Dosage Modifications for Non-hematologic Adverse Reactions
Dosage modifications for infusion-related reactions and other adverse drug reactions, including elevated liver function tests (LFTs), are described in Table 3. Monitor liver function tests prior to administration of RYTELO, weekly for the first cycle, prior to each cycle thereafter, and as clinically indicated.
| Adverse Reaction | Severity Grade Severity based on National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03. | Occurrence | Treatment Modification |
|---|---|---|---|
| Abbreviation: LFT = liver function test | |||
| Infusion-Related Reactions [see Warnings and Precautions (5.3) ] | Grade 2 or 3 | First and Second | Interrupt the RYTELO infusion until resolution of the adverse reaction or until the intensity of the reaction decreases to Grade 1; restart infusion at 50% of the infusion rate administered prior to the adverse reaction. |
| Third | For Grade 2, stop infusion. May restart at next cycle. For Grade 3, permanently discontinue RYTELO. | ||
| Grade 4 | First | Stop infusion, administer supportive care as appropriate and permanently discontinue RYTELO. | |
| Other adverse reactions including elevated LFTs [see Adverse Reactions (6.1) ] | Grade 3 or 4 | First and Second | Delay RYTELO until recovery of adverse reactions to Grade 1 or baseline; restart at one dose level lower. |
| Third | Permanently discontinue RYTELO. | ||
Preparation and Administration
RYTELO is provided as a lyophilized powder in a single-dose vial for intravenous infusion only and must be reconstituted and diluted prior to administration.
Use aseptic technique to prepare RYTELO.
RYTELO does not contain a preservative.
Reconstitution:
- Calculate the dose of RYTELO needed based on the patient's body weight (kg).
- Determine the number of RYTELO vials needed to achieve the required dose (total mg) per Table 4. More than one vial may be needed to achieve a full dose.
- Remove the RYTELO vials from the refrigerator and allow the vials to sit for 10 minutes to 15 minutes (not to exceed 30 minutes) to adjust to room temperature 20°C to 25°C (68°F to 77°F) before use.
- Reconstitute each vial of RYTELO with the volume of 0.9% Sodium Chloride Injection provided in Table 4 directly onto the lyophilized powder to obtain a concentration of 31.4 mg/mL of imetelstat.
| Strength Recommended to use the appropriate combination of vial strengths to most closely match the intended dose based on the patient's weight. | Volume of 0.9% Sodium Chloride Injection for Reconstitution per Vial | Final Concentration of Reconstituted Solution per Vial | Deliverable Volume per Vial |
|---|---|---|---|
| 47 mg | 1.8 mL | 31.4 mg/mL Each vial contains an overfill to account for loss of liquid during preparation and extraction of the reconstituted solution, resulting in the final concentration. | 1.5 mL |
| 188 mg | 6.3 mL | 31.4 mg/mL | 6 mL |
- Swirl each vial gently to avoid foaming until the powder is fully reconstituted (not to exceed 15 minutes). Do not shake.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The reconstituted solution in each vial should appear as a clear to slightly hazy solution, essentially free of visible contaminants, particles and/or particulates. Do not use if discoloration or particulate matter is present.
- Use the reconstituted solution immediately to prepare the RYTELO diluted solution in the infusion bag.
Dilution:
- Calculate the required volume of the reconstituted RYTELO solution needed to obtain the appropriate dose according to the patient's body weight.
- Withdraw a volume equal to the required reconstituted RYTELO solution from a 500 mL infusion bag of 0.9% Sodium Chloride Injection and discard it.
- Add the required volume of reconstituted RYTELO solution into the infusion bag so that the total final volume of RYTELO solution in the bag is approximately 500 mL. Discard any unused portion of the reconstituted solution remaining in each vial.
- Gently invert the infusion bag at least 5 times to ensure that the reconstituted RYTELO is well-mixed. Do not shake the infusion bag prior to administration.
Diluted RYTELO Solution Storage:
- If not used immediately, ensure that diluted solution for infusion is used within the total timeframes specified below, according to storage temperature:
- When stored at room temperature 20°C to 25°C (68°F to 77°F) : The total time from the reconstitution of RYTELO to completion of the intravenous infusion should not exceed 18 hours from the time of reconstitution.
- When stored refrigerated 2°C to 8°C (36°F to 46°F) : The total time from the reconstitution of RYTELO to completion of the intravenous infusion should not exceed 48 hours from the time of reconstitution.
Administration :
- Administer the diluted RYTELO solution by intravenous infusion only over a period of 2 hours.
DOSAGE FORMS AND STRENGTHS
- For injection: 47 mg of imetelstat supplied as a white to off-white or slightly yellow lyophilized powder in a single-dose vial for reconstitution.
- For injection: 188 mg of imetelstat supplied as a white to off-white or slightly yellow lyophilized powder in a single-dose vial for reconstitution.
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2 )
Pregnancy
Risk Summary
Based on findings in animal studies, RYTELO can cause embryo-fetal harm when administered to a pregnant woman. There are no available data on RYTELO use in pregnant women to evaluate for drug-associated risk. In embryo-fetal developmental toxicity studies, administration of imetelstat to pregnant mice and rabbits during the period of organogenesis resulted in embryo-fetal mortality, which in mice occurred at maternal exposures approximately 2.5 times the human exposure at the recommended clinical dose. Advise pregnant women of the potential risk to a fetus [see Data ] .
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In embryo-fetal developmental toxicity studies, imetelstat was administered by IV bolus injection at doses of 4.7, 9.4, 14.1 or 28.2 mg/kg/day on gestation days 6, 9, and 12 in mice, or by 2-hour intravenous infusion at doses of 4.7, 14.1, or 28.2 mg/kg on gestation days 6 and 13 in rabbits. In rabbits, the dose of 28.2 mg/kg was maternally toxic. Increased post-implantation loss due to an increase in early resorptions, resulting in a decrease in viable fetuses and litter was noted in mice at 28.2 mg/kg and in rabbits starting at 14.1 mg/kg; corresponding to exposures (based on AUC) that are approximately 2.5-times (mice) or 9.3-times (rabbits) the human exposure at the recommended clinical dose.
Lactation
Risk Summary
There is no data on the presence of imetelstat in human milk, or the effects RYTELO on the breastfed child, or milk production. Because of the potential for adverse reactions in breastfed children, advise women not to breastfeed during treatment with RYTELO and for 1 week after the last dose.
Females and Males of Reproductive Potential
Based on findings from animal studies, RYTELO can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1) ] .
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating treatment with RYTELO.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with RYTELO and for 1 week after the last dose [see Use in Specific Populations (8.1) ] .
Infertility
Females
Based on findings in animals, RYTELO may impair fertility in females of reproductive potential. The effect on fertility is reversible [see Nonclinical Toxicology (13.1) ] .
Pediatric Use
Safety and effectiveness of RYTELO in pediatric patients have not been established.
Geriatric Use
Of the 118 patients with low- to intermediate-1 risk myelodysplastic syndromes (MDS) in the clinical trial who received RYTELO, 91 (77.1%) patients were 65 years of age and older and 35 (29.7%) patients 75 years of age and older. No differences in safety or efficacy were observed between older (≥ 65 years) and younger patients.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
- Thrombocytopenia : Grade 3 and Grade 4 thrombocytopenia occurred; obtain complete blood cell counts prior to initiation of RYTELO, weekly for the first two cycles, and prior to each cycle thereafter to monitor. Delay or dose reduce as recommended. (2.3 , 5.1 )
- Neutropenia : Grade 3 and Grade 4 neutropenia occurred; obtain complete blood cell counts prior to initiation of RYTELO, weekly for the first two cycles, and prior to each cycle thereafter to monitor. Delay or dose reduce as recommended. (2.3 , 5.2 )
- Infusion-Related Reactions : Premedicate before infusion. Interrupt, decrease the rate of infusion, or permanently discontinue RYTELO based on severity. (2.2 , 2.3 , 5.3 )
- Embryo-Fetal Toxicity : Can cause embryo-fetal harm. Advise females of reproductive potential of potential risk to a fetus and to use effective contraception. (5.4 , 8.1 , 8.3 )
Thrombocytopenia
RYTELO can cause thrombocytopenia based on laboratory values. In the clinical trial, new or worsening Grade 3 or 4 decreased platelets occurred in 65% of patients with MDS treated with RYTELO [see Adverse Reactions (6.1) ] .
Median time to onset of first occurrence of Grade 3 or 4 decreased platelets was 6 weeks (range: 2 to 88 weeks) and median time to recovery from each occurrence of Grade 3 or 4 decreased platelets to Grade 2 or lower, or last value available, was 1.3 weeks (range: 0.1 to 13 weeks). Grade 3 or 4 decreased platelets occurred throughout treatment with RYTELO, with 48% of patients experiencing Grade 3 or Grade 4 thrombocytopenia during cycles 1-3, 31% during cycles 4-6, 33% during cycles 7-12, and 24% during cycles 13 and beyond. Grade 3 or 4 bleeding was seen in 2.5% of patients, including gastrointestinal bleeding (1.7%) and hematuria (0.8%).
Monitor patients with thrombocytopenia for bleeding.
Monitor complete blood cell counts prior to initiation of RYTELO, weekly for the first two cycles, prior to each cycle thereafter, and as clinically indicated. Administer platelet transfusions as appropriate. Delay the next cycle and resume at the same or reduced dose, or discontinue as recommended [see Dosage and Administration (2.3) ] .
Neutropenia
RYTELO can cause neutropenia based on laboratory values. In the clinical trial, new or worsening Grade 3 or 4 decreased neutrophils occurred in 72% of patients with MDS treated with RYTELO [see Adverse Reactions (6.1) ] .
Median time to onset of first occurrence of Grade 3 or 4 decreased neutrophils was 4.6 weeks (range: 1 to 81 weeks) and median time to recovery from each occurrence of Grade 3 or 4 decreased neutrophils to Grade 2 or lower, or last value available, was 1.9 weeks (range: 0 to 16 weeks). Grade 3 or 4 decreased neutrophils occurred throughout treatment with RYTELO, with 65% of patients experiencing Grade 3 or Grade 4 neutropenia during cycles 1-3, 35% during cycles 4-6, 32% during cycles 7-12, and 39% during cycles 13 and beyond. Febrile neutropenia occurred in 0.8% and sepsis in 4.2%.
Monitor patients with Grade 3 or 4 neutropenia for infections, including sepsis.
Monitor complete blood cell counts prior to initiation of RYTELO, weekly for the first two cycles, prior to each cycle thereafter, and as clinically indicated. Administer growth factors and anti-infective therapies for treatment or prophylaxis as appropriate. Delay the next cycle and resume at the same or reduced dose, or discontinue as recommended [see Dosage and Administration (2.3) ] .
Infusion-Related Reactions
RYTELO can cause infusion-related reactions. In the clinical trial, infusion-related reactions occurred in 8% of patients with MDS treated with RYTELO; Grade 3 or 4 infusion-related reactions occurred in 1.7%, including hypertensive crisis (0.8%). The most common infusion-related reaction was headache (4.2%). Infusion-related reactions usually occur during or shortly after the end of the infusion.
Premedicate patients at least 30 minutes prior to infusion with diphenhydramine and hydrocortisone as recommended and monitor patients for at least one hour following the infusion as recommended [see Dosage and Administration (2.2) ]. Manage symptoms of infusion-related reactions with supportive care and infusion interruptions, decrease infusion rate, or permanently discontinue as recommended [see Dosage and Administration (2.3) ] .
Embryo-Fetal Toxicity
Based on findings in animals, RYTELO can cause embryo-fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of imetelstat to pregnant mice during the period of organogenesis resulted in embryo-fetal mortality at maternal exposures (AUC) 2.5-times the human exposure at the recommended clinical dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with RYTELO and for 1 week after the last dose [see Use in Specific Populations (8.1 , 8.3) ] .
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Thrombocytopenia [see Warnings and Precautions (5.1) ]
- Neutropenia [see Warnings and Precautions (5.2) ]
- Infusion-Related Reactions [see Warnings and Precautions (5.3) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Low- to Intermediate-Risk Myelodysplastic Syndromes
The safety of RYTELO was evaluated in a randomized, double-blind, placebo-controlled, multicenter trial (IMerge) in 177 adult patients with International Prognostic Scoring System (IPSS) low- to intermediate-1 risk MDS who were transfusion-dependent and relapsed or refractory to or ineligible for ESA treatment [see Clinical Studies (14) ] . The safety population included patients who received at least one dose of either RYTELO (n=118) or placebo (n=59) at 7.1 mg/kg as an intravenous infusion administered over two hours every 4 weeks. The median time on treatment with RYTELO was 8 months (range, 0 to 38 months); 69% of patients were exposed to RYTELO for 24 weeks or longer and 45% were exposed for 48 weeks or longer.
The median age of patients who received at least one dose of RYTELO was 72 years (range: 44 to 87 years) with 77% of patients 65 years of age and older and 30% of patients 75 years of age and older. Participants were 60% male, 81% White, 7% Asian, and 0.8% Black.
Serious adverse reactions occurred in 32% of patients who received RYTELO. Serious adverse reactions in > 2% of patients included sepsis (4.2%), fracture (3.4%), cardiac failure (2.5%), and hemorrhage (2.5%). Fatal adverse reactions occurred in 0.8% of patients who received RYTELO, including sepsis (0.8%).
Permanent discontinuation of RYTELO due to an adverse reaction occurred in 15% of patients. Adverse reactions which resulted in permanent discontinuation of RYTELO in > 2% of patients included neutropenia and thrombocytopenia.
Dosage interruptions of RYTELO due to an adverse reaction occurred in 80% of patients. Adverse reactions which required dosage interruption in > 5% of patients included neutropenia, thrombocytopenia and infections.
Dose reductions of RYTELO due to an adverse reaction occurred in 49% of patients. Adverse reactions which required dose reductions in > 2% of patients included neutropenia and thrombocytopenia.
The most common (≥10% with a difference between arms of >5% compared to placebo) adverse reactions, including laboratory abnormalities, were decreased platelets, decreased white blood cells, decreased neutrophils, increased AST, increased alkaline phosphatase, increased ALT, fatigue, prolonged partial thromboplastin time, arthralgia/myalgia, COVID-19 infections, and headache.
Table 5 summarizes the adverse reactions in IMerge.
| Adverse Reaction | RYTELO (N=118) | Placebo (N=59) | ||
|---|---|---|---|---|
| All Grades % | Grades 3 or 4 % | All Grades % | Grades 3 or 4 % | |
| Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03. | ||||
| General disorders and administrative site conditions | ||||
| Fatigue Fatigue: asthenia, fatigue, and malaise. | 29 | 0 | 20 | 3.4 |
| Musculoskeletal and connective tissue disorders | ||||
| Arthralgia/myalgia Arthralgia/myalgia: arthralgia, back pain, bone pain, musculoskeletal pain, myalgia, neck pain, non-cardiac chest pain, pain, pain in extremity, pain in jaw, and pelvic pain. | 25 | 2.5 | 19 | 5 |
| Infections and infestations | ||||
| COVID-19 COVID-19: asymptomatic COVID-19, COVID-19, COVID-19 pneumonia, and SARS-CoV-2 antibody test positive. | 19 | 1.7 | 14 | 5 |
| Urinary tract infection Urinary tract infection: cystitis, Escherichia urinary tract infection, renal abscess, and urinary tract infection. | 9 | 2.5 | 7 | 0 |
| Nervous system disorders | ||||
| Headache | 13 | 0.8 | 5 | 0 |
| Syncope Syncope: fall, pre-syncope, and syncope. | 7 | 1.7 | 1.7 | 0 |
| Immune system disorders | ||||
| Infusion-related reactions Infusion-related reactions: abdominal pain, arthralgia, asthenia, back pain, bone pain, diarrhea, erythema, headache, hypertensive crisis, malaise, non-cardiac chest pain, pruritus, and urticaria. Only events considered related to infusion-related reactions are included. | 8 | 1.7 | 3.4 | 0 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Epistaxis | 7 | 0 | 0 | 0 |
| Vascular disorders | ||||
| Hematoma | 6 | 0 | 0 | 0 |
| Skin and subcutaneous tissue disorders | ||||
| Pruritus | 6 | 0 | 1.7 | 0 |
| Cardiac disorders | ||||
| Atrial arrhythmia Atrial arrhythmia: atrial fibrillation and atrial flutter. | 6 | 1.7 | 3.4 | 1.7 |
| Injury, poisoning and procedural complications | ||||
| Fractures Fractures: femur fracture, hand fracture, hip fracture, humerus fracture, lumbar vertebral fracture, and thoracic vertebral fracture. | 5 | 3.4 | 1.7 | 1.7 |
Clinically relevant adverse reactions in < 5% of patients who received RYTELO included febrile neutropenia, sepsis, gastrointestinal hemorrhage, and hypertension.
Table 6 summarizes the laboratory abnormalities in IMerge.
| Laboratory Abnormality | RYTELO The denominator used to calculate the rate varied from 97 to 118 based on the number of patients with a baseline value and at least one post-treatment value. | Placebo The denominator used to calculate the rate varied from 50 to 59 based on the number of patients with a baseline value and at least one post-treatment value. | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | |
| Graded according to National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03. ALP = alkaline phosphatase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; PTT = partial thromboplastin time | ||||
| Hematology | ||||
| Platelet count decreased | 97 | 65 | 34 | 8 |
| White blood cell count decreased | 94 | 53 | 59 | 1.7 |
| Neutrophil count decreased | 92 | 72 | 47 | 7 |
| PTT prolonged | 26 | 1 | 18 | 4 |
| Chemistry | ||||
| AST increased | 53 | 0.8 | 22 | 1.7 |
| ALP increased | 48 | 0 | 12 | 0 |
| ALT increased | 43 | 3.4 | 37 | 5 |
DESCRIPTION
RYTELO for injection contains imetelstat, an oligonucleotide telomerase inhibitor for intravenous use. Imetelstat sodium is a white to off-white or slightly yellow, amorphous, solid powder. It is highly soluble in aqueous solutions, including in 0.9% Sodium Chloride Injection, at both refrigerated and room temperatures. Imetelstat sodium is hygroscopic.
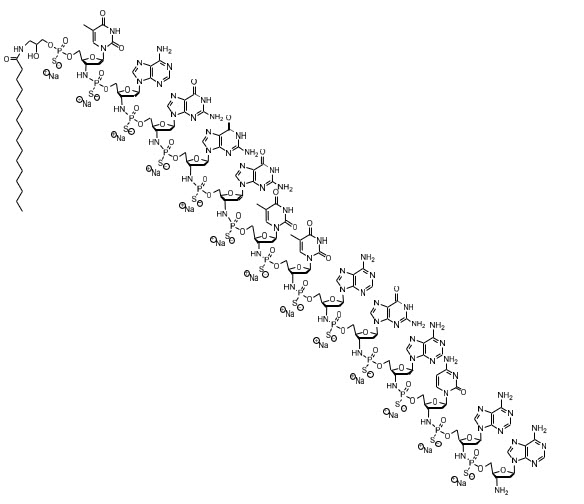
The chemical name for the imetelstat sodium drug substance is DNA, d(3'-amino-3'-deoxy-P-thio) (T-A-G-G-G-T-T-A-G-A-C-A-A), 5'-[ O -[2-hydroxy-3-(hexadecanoylamino)propyl] phosphorothioate], sodium salt (1:13). The molecular formula is C 148 H 198 N 68 O 53 P 13 S 13 Na 13 (as sodium salt), which equates to a formula weight of 4896 g/mol. The molecular formula for the free acid form is C 148 H 211 N 68 O 53 P 13 S 13 which equates to a formula weight of 4610 g/mol. The structural formula for imetelstat sodium is:
RYTELO (imetelstat) for injection is a sterile, preservative-free, white to off-white or slightly yellow lyophilized powder for intravenous infusion after reconstitution and dilution. Each single-dose vial provides either 47 mg of imetelstat (equivalent to 50 mg imetelstat sodium) or 188 mg of imetelstat (equivalent to 200 mg imetelstat sodium). The following inactive ingredients may be added during manufacturing: sodium carbonate anhydrous (for the 47 mg preparation) / sodium carbonate monohydrate (for the 188 mg preparation) or hydrochloric acid (to adjust to pH of 7.0 to 8.5).
CLINICAL PHARMACOLOGY
Mechanism of Action
Imetelstat is an oligonucleotide human telomerase inhibitor that binds to the template region of the RNA component of human telomerase (hTR), inhibits telomerase enzymatic activity and prevents telomere binding.
Increased telomerase activity and human telomerase reverse transcriptase (hTERT) RNA expression have been reported in MDS and malignant stem and progenitor cells. Nonclinical studies showed imetelstat treatment led to reduction of telomere length, reduction of malignant stem and progenitor cell proliferation, and induction of apoptotic cell death.
Pharmacodynamics
Grade 3 and 4 Thrombocytopenia
Higher imetelstat exposure is associated with higher incidence of Grade 3 and 4 thrombocytopenia in patients with MDS.
At the maximum recommended dose, clinically significant QTc interval prolongation was not observed.
Pharmacokinetics
Imetelstat plasma geometric mean (coefficient of variation [CV] %) maximum concentration (C max ) is 18.3 µM (27.3%) and the area under the concentration-time curve from time 0 to 28 days (AUC 0-28 ) was 114.2 h•µM (43.2%). Imetelstat does not accumulate between treatment cycles.
Distribution
Following a single intravenous dose of 7.1 mg/kg of RYTELO administered over 2 hours in patients with MDS, the geometric mean (CV%) volume of distribution is approximately 14.1 L (27.2%). In vitro human plasma protein binding is greater than 94%.
Elimination
The imetelstat geometric mean (CV%) apparent plasma half-life is approximately 4.9 hours (43.2%) at the approved recommended dosage.
Metabolism
Imetelstat is expected to be metabolized by nucleases to nucleotides of various lengths.
Specific populations
No clinically significant differences in the pharmacokinetics of imetelstat were observed based on age (21 to 87 years), sex, race (81% White, 4% Asian, 7% Black, 8% other/unknown), mild to moderate renal impairment (CL cr 30 to < 90 mL/min), mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN, or total bilirubin > 1× to 1.5× ULN and any AST), or moderate hepatic impairment (total bilirubin > 1.5× to 3× ULN and any AST). The effect of severe renal impairment (CL cr 15 to < 30 mL/min), end-stage renal disease, or severe hepatic impairment (total bilirubin > 3× ULN and any AST) has not been established.
Drug Interaction Studies
In Vitro Studies
CYP Enzymes : Imetelstat is not an inhibitor or inducer of CYP450 enzymes.
Transporter systems : Imetelstat is an inhibitor of OATP1B1 and OATP1B3.
Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of RYTELO.
Seventeen percent (28/166) of evaluable patients with low- or intermediate-1 risk MDS tested positive for imetelstat anti-drug antibodies, with a median onset of 38 weeks following the study drug dosage of RYTELO for a median duration of treatment of 35 weeks in Phase 2 and Phase 3 of the IMerge study. There was no clinically significant effect of ADA on the PK, safety, or efficacy of RYTELO among the study participants who developed anti-drug antibodies.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with imetelstat.
In vitro, imetelstat was not mutagenic in the bacterial mutagenicity assay (Ames test) or clastogenic in the chromosomal aberrations assay using cultured human peripheral blood lymphocytes. Imetelstat was not genotoxic in an in vivo mouse micronucleus assay at intravenous dose levels up to approximately 104 mg/kg.
Fertility studies have not been conducted with imetelstat. Female monkeys given 14.1 mg/kg once weekly for 9 months exhibited uterine endometrial atrophy in a general toxicology study. This effect was observed at a mean exposure (based on AUC) that is approximately 14.4-times the human exposure at the recommended clinical dose. This finding was not present in animals following a 14-week recovery period.
CLINICAL STUDIES
Myelodysplastic Syndromes (MDS)
The efficacy of RYTELO was evaluated in a randomized, double-blind, placebo-controlled, multicenter trial (IMerge; NCT02598661) in 178 patients enrolled with International Prognostic Scoring System (IPSS) low- or intermediate-1 risk MDS who were transfusion-dependent (requiring ≥ 4 red blood cell (RBC) units over an 8-week period during the 16 weeks prior to randomization). Eligible patients were required to have failed to respond or have lost response or be ineligible for erythropoiesis-stimulating agents (ESAs); and had an absolute neutrophil count of 1.5 × 10 9 /L or greater and platelets 75 × 10 9 /L or greater. Patients were ineligible if they had del(5q) cytogenetic abnormality or had received prior treatment with lenalidomide or hypomethylating agents.
Participants were randomized in a 2:1 ratio to receive an intravenous infusion of RYTELO (n=118) 7.1 mg/kg or placebo (n=60) in 28-day treatment cycles until disease progression, unacceptable toxicity, or withdrawal from the study. Randomization was stratified by prior RBC transfusion burden and by IPSS risk group. All patients received supportive care, which included RBC transfusions.
Of the 178 patients enrolled, the median age was 72 years (range: 39 to 87 years), with 62% male, and 80% White, 6% Asian, 1.7% Black, 12% other or not reported. The key baseline disease characteristics of the efficacy population are shown in Table 7.
| Disease Characteristic | RYTELO (N=118) | Placebo (N=60) |
|---|---|---|
| Abbreviations: ECOG = Eastern Cooperative Oncology Group; ESA = erythropoietin stimulating agents; IPSS = International Prognostic Scoring System; RBC = Red Blood Cell; MDS = myelodysplastic syndromes; sEPO = serum erythropoietin; WHO = World Health Organization | ||
| Time since original diagnosis | ||
| Median years (range) | 3.5 (0.1, 26.7) | 2.8 (0.2, 25.7) |
| ECOG Score (0, 1, 2), n (%) | ||
| 0: Asymptomatic | 42 (35.6) | 21 (35) |
| 1: Symptomatic fully ambulatory | 70 (59.3) | 39 (65) |
| 2: Symptomatic in bed less than 50% of the day | 6 (5.1) | 0 |
| IPSS Risk Classification, n (%) | ||
| Low | 80 (67.8) | 39 (65) |
| Intermediate-1 | 38 (32.2) | 21 (35) |
| Prior RBC transfusion burden Prior RBC transfusion burden is defined as the maximum number of RBC units transfused over an 8-week period during the 16 weeks prior to randomization. , n (%) | ||
| 4 to 6 units | 62 (52.5) | 33 (55) |
| > 6 units | 56 (47.5) | 27 (45) |
| WHO Classification (2008), n (%) | ||
| RS+ RS+ includes refractory anemia with ring sideroblasts (RARS)/refractory cytopenia with multilineage dysplasia and ≥ 15% ringed sideroblasts (RCMD-RS). | 73 (61.9) | 37 (61.7) |
| RS˗ RS˗ includes others. | 44 (37.3) | 23 (38.3) |
| Missing | 1 (0.8) | 0 |
| Baseline sEPO, n (%) | ||
| ≤ 500 mU/mL | 87 (73.7) | 36 (60) |
| > 500 mU/mL | 26 (22) | 22 (36.7) |
| Missing | 5 (4.2) | 2 (3.3) |
| Prior ESA Use, n (%) | ||
| Yes | 108 (91.5) | 52 (86.7) |
| No | 10 (8.5) | 8 (13.3) |
| Baseline blood counts, median | ||
| Neutrophils (cells/L) | 2.6 × 10 9 | 2.7 × 10 9 |
| Hemoglobin (g/dL) | 7.9 | 7.8 |
| Platelets (cells/L) | 230 × 10 9 | 230 × 10 9 |
Efficacy was established after a median follow up time of 19.5 months (range: 1.4 to 36.2) in the imetelstat group and 17.5 months (range: 0.7 to 34.3) in the placebo group based upon the proportion of patients who achieved ≥ 8-week and ≥ 24-week RBC-TI, defined as the absence of RBC transfusion(s) during any consecutive 8-week (56-day) period, and during any consecutive 24-week (168-day) period, respectively, from randomization until the start of subsequent anti-cancer therapy (if any). The efficacy results are summarized in Table 8.
| RYTELO (N=118) | Placebo (N=60) | % Difference (95% CI) 95% CI based on Wilson Score method. | p -value p- value is based on Cochran-Mantel-Haenszel (CMH) controlling for prior RBC transfusion burden (≤ 6 versus >6 units RBC) and IPSS risk group (low versus intermediate-1) applied to randomization. | |
|---|---|---|---|---|
| Abbreviations: CI = Confidence Interval; RBC = Red Blood Cell; TI = Transfusion Independence | ||||
| Rate of ≥ 8-week RBC TI | ||||
| ≥ 8-week RBC TI, n (%) | 47 (39.8) | 9 (15.0) | 24.8 (9.9, 36.9) | < 0.001 |
| 95% CI for response rate (%) Exact Clopper-Pearson confidence interval. | (30.9, 49.3) | (7.1, 26.6) | ||
| Rate of ≥ 24-week RBC TI | ||||
| ≥ 24-week RBC TI, n (%) | 33 (28.0) | 2 (3.3) | 24.6 | |
| 95% CI for response rate (%) | (20.1, 37.0) | (0.4, 11.5) | (12.6, 34.2) | < 0.001 |
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
RYTELO (imetelstat) for injection is a preservative free, white to off-white or slightly yellow, lyophilized powder available as:
| Carton Contents | NDC |
|---|---|
| One RYTELO 47 mg single-dose vial | 82959-112-01 |
| One RYTELO 188 mg single-dose vial | 82959-111-01 |
Storage
Store vials refrigerated at 2°C to 8°C (36°F to 46°F) in original carton.
Do not freeze.
Mechanism of Action
Imetelstat is an oligonucleotide human telomerase inhibitor that binds to the template region of the RNA component of human telomerase (hTR), inhibits telomerase enzymatic activity and prevents telomere binding.
Increased telomerase activity and human telomerase reverse transcriptase (hTERT) RNA expression have been reported in MDS and malignant stem and progenitor cells. Nonclinical studies showed imetelstat treatment led to reduction of telomere length, reduction of malignant stem and progenitor cell proliferation, and induction of apoptotic cell death.