Get your patient on Romvimza - Vimseltinib capsule (Vimseltinib)
Romvimza - Vimseltinib capsule prescribing information
INDICATIONS AND USAGE
ROMVIMZA is indicated for treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) for which surgical resection will potentially cause worsening functional limitation or severe morbidity.
DOSAGE AND ADMINISTRATION
Recommended Dosage
The recommended dosage of ROMVIMZA is 30 mg orally taken twice weekly, with a minimum of 72 hours between doses, as directed on the blister package [ see Clinical Pharmacology (12.3 ) ]. Instruct patients to follow the schedule on the blister package and to take ROMVIMZA on the same days each week.
- ROMVIMZA may be taken with or without food.
- Swallow ROMVIMZA capsules whole. Do not open, break, or chew the capsules.
- If a dose is missed by 48 hours or less, take the missed dose as soon as possible and take the next dose on its regularly scheduled day. If a dose is missed by more than 48 hours, skip the missed dose, and take the next dose on its regularly scheduled day.
- If vomiting occurs within 30 minutes of taking a dose, repeat that dose. Otherwise, take the next dose on its regularly scheduled day.
Dose Modifications for Adverse Reactions
The recommended dose reductions for adverse reactions are provided in Table 1 .
| Dose Reduction | Twice Weekly Dose |
| First | 20 mg |
| Second | 14 mg |
Permanently discontinue ROMVIMZA in patients who are unable to tolerate 14 mg orally twice weekly.
The recommended dosage modifications for hepatotoxicity are summarized in Table 2 .
| Hepatotoxicity Severity | ROMVIMZA Dosage Modifications |
|---|---|
ALT = alanine aminotransferase; ALP = alkaline phosphatase; AST = aspartate aminotransferase; INR = International normalized ratio; ULN = upper limit of normal | |
| AST and/or ALT increases >3–5 times ULN and total bilirubin increases up to 2 times ULN | Withhold ROMVIMZA until AST and ALT resolve to baseline or ≤3 times ULN, and bilirubin resolves to baseline. Resume at the next lower dose level once Hy's law has been definitively ruled out. Permanently discontinue if adverse reaction does not resolve within 4 weeks. |
| OR | |
| Total bilirubin increases up to 2 times ULN | |
| AST and/or ALT increases >3–5 times ULN, and total bilirubin increases >2 times ULN or INR >1.5 and ALP <2 times ULN | Withhold ROMVIMZA until AST and ALT resolve to baseline or ≤3 times ULN, and bilirubin resolves to baseline. Resume at the next lower dose level once Hy's law has been definitively ruled out. Permanently discontinue if adverse reaction does not resolve within 4 weeks. |
| OR | |
| Total bilirubin increases >2 times ULN | |
| AST and/or ALT increases >5–8 times ULN, and total bilirubin ≤ULN and without clinical symptoms | Withhold ROMVIMZA until AST and ALT resolve to ≤3 times ULN or baseline. Permanently discontinue if adverse reaction does not resolve within 4 weeks. |
| AST and/or ALT increases >5-8 times ULN and total bilirubin increase >ULN, or INR >1.5, or ALP >2 times ULN | Permanently discontinue ROMVIMZA. |
| AST and/or ALT increases >8 times ULN | Permanently discontinue ROMVIMZA. |
Dosage Modification for P-glycoprotein (P-gp) Substrates
Avoid concomitant use of ROMVIMZA with P-gp substrates. If concomitant use of a P-gp substrate is unavoidable, administer ROMVIMZA at least 4 hours before taking the P-gp substrate unless otherwise recommended in the substrate Prescribing Information [ see Drug Interactions (7.1 ) ].
DOSAGE FORMS AND STRENGTHS
14 mg capsule
Orange cap, white body size 4 capsule imprinted with “DCV14” in black ink.
20 mg capsule
Yellow cap, white body size 2 capsule imprinted with “DCV20” in black ink.
30 mg capsule
Light blue cap, white body size 1 capsule imprinted with “DCV30” in black ink.
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2 )
Pregnancy
Risk Summary
Based on data from animal studies and its mechanism of action, ROMVIMZA can cause fetal harm when administered to a pregnant woman. There are no available data on vimseltinib use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In female rats administered vimseltinib during the period of organogenesis, fetal structural abnormalities occurred at exposures that were at least 3 times the recommended dose based on AUC ( see Data ) . Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In a rat embryo-fetal development study, pregnant female rats were dosed once daily during the period of organogenesis (gestational days 6 to 17) at doses of 2.5, 5, or 15 mg/kg/day. Structural abnormalities (skeletal variations) occurred at ≥2.5 mg/kg/day (approximately 3 times the exposure at the recommended dose based on AUC). Additional structural abnormalities (cardiac malformations) were observed at the highest dose of 15 mg/kg/day (approximately 23 times the exposure at the recommended dose based on AUC).
Lactation
Risk Summary
There are no data on the presence of vimseltinib or its metabolites in either human or animal milk or its effects on a breastfed child or on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with ROMVIMZA and for 1 month after the last dose.
Females and Males of Reproductive Potential
ROMVIMZA can cause fetal harm when administered to a pregnant woman [ see Use in Specific Populations (8.1 ) ].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to the initiation of ROMVIMZA [ see Use in Specific Populations (8.1 ) ].
Contraception
Infertility
Females and Males
Based on findings from animal studies, ROMVIMZA may impair fertility [ see Nonclinical Toxicology (13.1 ) ].
Pediatric Use
The safety and effectiveness of ROMVIMZA in pediatric patients have not been established.
Animal Toxicity Data
In a 26-week repeat-dose toxicology study, rats administered vimseltinib at ≥2.5 mg/kg/day had physeal thickening and decay of the incisors and molars. Bone and tooth toxicities occurred at exposures at least 8 times the recommended dose based on AUC.
Geriatric Use
Clinical studies of ROMVIMZA did not include a sufficient number of patients aged 65 years and older to determine whether they respond differently from younger patients.
Hepatic Impairment
No dose adjustment is recommended for patients with mild (bilirubin ≤upper limit of normal (ULN) and aspartate aminotransferase (AST) >ULN or bilirubin >1x to 1.5x ULN and any AST) hepatic impairment. ROMVIMZA has not been studied in patients with moderate (bilirubin >1.5x to 3x ULN and any AST) or severe (bilirubin >3x ULN and any AST) hepatic impairment [see Clinical Pharmacology (12.3 )] .
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Hepatotoxicity: Elevated AST and ALT can occur. Evaluate liver tests prior to initiation of treatment and during treatment. (2.2 , 5.1 )
Embryo-fetal toxicity: Can cause fetal harm. Advise patients of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.2 , 8.1 , 8.3 )
Allergic Reactions to FD&C Yellow No. 5 (tartrazine) and No. 6 (Sunset Yellow FCF): 14 mg capsule contains FD&C Yellow No. 6 (Sunset Yellow FCF); 20 mg capsule contains FD&C Yellow No.5 (tartrazine) and No. 6 (Sunset Yellow FCF) as color additives, which may cause allergic reactions (including bronchial asthma) in certain susceptible patients. (5.3 )
Increased serum creatinine without affecting renal function: Increases in serum creatinine can occur. Use alternative measures that are not based on serum creatinine to assess renal function. (5.4 )
Hepatotoxicity
Cases of serious and fatal liver injury have occurred with the use of another kinase inhibitor that targets CSF1R [see Clinical Pharmacology (12.1 )]. Serious and fatal liver injury have not been observed with ROMVIMZA.
Across clinical trials in 253 patients treated with ROMVIMZA, 2% had Grade 3 increased AST, and 1% had Grade 3 increased ALT. Dose interruptions occurred in 2% of patients and dose reductions occurred in 1% of patients due to AST/ALT increase. One patient discontinued therapy due to Grade 3 AST increased.
Avoid ROMVIMZA in patients with pre-existing increased serum transaminases; total bilirubin or direct bilirubin (>ULN); or active liver or biliary tract disease, including ALP. Monitor liver tests, including AST, ALT, total bilirubin, direct bilirubin, ALP and gamma-glutamyl transferase (GGT), prior to initiation of ROMVIMZA, twice a month for the first two months and once every 3 months for the first year of therapy and as clinically indicated thereafter. Withhold and reduce the dose, or permanently discontinue ROMVIMZA based on the severity of the hepatotoxicity [ see Dosage and Administration (2.2 ) ].
Embryo-Fetal Toxicity
Based on data from animal studies and its mechanism of action, ROMVIMZA can cause fetal harm when administered to pregnant women. In female rats administered vimseltinib, fetal structural abnormalities occurred at exposures that were at least 3 times the recommended dose based on area under the curve (AUC). Advise pregnant women on the potential risk to the fetus.
Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with ROMVIMZA and for 1 month after the last dose [ see Use in Specific Populations (8.1 , 8.3 ) ].
Allergic Reactions to FD&C Yellow No.5 (Tartrazine) and No. 6 (Sunset Yellow FCF)
ROMVIMZA 20 mg capsule contains FD&C Yellow No. 5 (tartrazine) which may cause allergic reactions (including bronchial asthma) in certain susceptible patients. Although the overall incidence of FD&C Yellow No. 5 (tartrazine) sensitivity in the general population is low, it is frequently seen in patients who also have aspirin sensitivity.
ROMVIMZA 14 mg and 20 mg capsules contain FD&C Yellow No.6 (Sunset Yellow FCF), which may cause allergic reactions.
Increased Creatinine without Affecting Renal Function
In MOTION, serum creatinine increased (mean increase of 19 μmol/L) and reached a maximum mean increase by 10.4 weeks compared to baseline. These increases in serum creatinine may not be associated with changes in renal function. Increases in creatinine reversed upon ROMVIMZA discontinuation. The increases in serum creatinine may be due to inhibition of renal tubular secretion transporters [ see Drug Interactions (7.1 ) and Clinical Pharmacology (12.3 ) ]. During ROMVIMZA treatment, use alternative measures that are not based on serum creatinine to assess renal function.
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hepatotoxicity [ see Warnings and Precautions (5.1 ) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the Warnings and Precautions reflects exposure to ROMVIMZA in 83 patients with TGCT enrolled in the double-blind portion and in 35 patients with TGCT in the open-label portion who crossed over to ROMVIMZA in MOTION, and in 135 patients with TGCT or solid tumors in other clinical trials.
The safety of ROMVIMZA was evaluated in 83 adult patients with TGCT in MOTION [ see Clinical Studies (14 ) ]. MOTION excluded patients with bilirubin, AST, or ALT >ULN. All patients received ROMVIMZA twice weekly until disease progression or unacceptable toxicity. Among these patients, 82% were exposed for 6 months or longer and 30% were exposed for greater than one year.
Serious adverse reactions occurred in 2.4% of patients who received ROMVIMZA. Serious adverse reactions in ≥1% included subcutaneous abscess (1.2%) and cellulitis (1.2%).
Permanent discontinuation due to an adverse reaction occurred in 4.8% of patients who received ROMVIMZA. Adverse reactions leading to permanent discontinuation in one patient each included periorbital edema, neuropathy, rash, and hypertension.
Dose reductions due to an adverse reaction or laboratory abnormality occurred in 39% of patients who received ROMVIMZA. Adverse reactions leading to dose reductions in ≥2% of patients receiving ROMVIMZA were rash, periorbital edema, peripheral edema, fatigue, pruritus, face edema, increased CPK, neuropathy, and hypertension.
Dose interruptions due to an adverse reaction or laboratory abnormality occurred in 40% of patients who received ROMVIMZA. Adverse reactions leading to interruptions in ≥2% of patients included rash, fatigue, peripheral edema, increased CPK, periorbital edema, face edema, pruritus, neuropathy, and hypertension.
The most common (≥20%) adverse reactions, including laboratory abnormalities that occurred in patients receiving ROMVIMZA were increased AST, periorbital edema, fatigue, rash, increased cholesterol, peripheral edema, face edema, decreased neutrophils, decreased leukocytes, pruritus, and increased ALT. Table 3 and Table 4 summarize the adverse reactions and laboratory abnormalities in MOTION during the randomized phase through Week 25.
| Adverse Reaction• | ROMVIMZA N=83 | Placebo N=39 | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | |
•The severity of adverse reactions was assessed using CTCAE v5.0. | ||||
1 Includes multiple related terms | ||||
| Eye disorders | ||||
| Periorbital edema 1 | 60 | 3.6 | 21 | 0 |
| Lacrimation increased | 12 | 0 | 0 | 0 |
| Dry eye 1 | 10 | 0 | 0 | 0 |
| General disorders and administration site conditions | ||||
| Fatigue 1 | 59 | 1.2 | 38 | 2.6 |
| Peripheral edema 1 | 33 | 1.2 | 8 | 0 |
| Face edema | 31 | 1.2 | 8 | 0 |
| Skin and subcutaneous tissue disorders | ||||
| Rash 1 | 47 | 3.6 | 5 | 0 |
| Pruritus | 29 | 2.4 | 8 | 0 |
| Vascular disorders | ||||
| Hypertension | 17 | 4.8 | 10 | 2.6 |
| Nervous system disorders | ||||
| Neuropathy 1 | 12 | 1.2 | 2.6 | 0 |
Other clinically significant adverse reactions occurring in <10% of patients treated with ROMVIMZA include blurred vision (6%).
| Laboratory Abnormality a | ROMVIMZA N=83 | Placebo N=39 b | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | |
AST – aspartate aminotransferase, ALT – alanine aminotransferase, ALP – alkaline phosphatase | ||||
a The severity of adverse reactions was assessed using CTCAE v5.0. | ||||
b The denominator used to calculate the rate was 83 for ROMVIMZA and 38 for placebo based on the number of patients with a baseline value and at least one post-treatment value. | ||||
| Chemistry | ||||
| AST increased | 92 | 0 | 11 | 0 |
| Cholesterol increased | 43 | 0 | 16 | 0 |
| ALT increased | 24 | 0 | 16 | 0 |
| Creatinine increased | 17 | 0 | 2.6 | 0 |
| ALP increased | 14 | 0 | 8 | 0 |
| Magnesium increased | 13 | 1.2 | 2.6 | 0 |
| Calcium decreased | 13 | 0 | 2.6 | 0 |
| Hematology | ||||
| Neutrophils decreased | 31 | 1.2 | 2.6 | 0 |
| Leukocytes decreased | 29 | 0 | 8 | 0 |
Additional clinically significant laboratory abnormality: Increased Creatine Phosphokinase (CPK)
DRUG INTERACTIONS
- P-glycoprotein (P-gp) substrates : Avoid concomitant use of ROMVIMZA with P-gp substrates. If concomitant use cannot be avoided, take ROMVIMZA at least 4 hours prior to P-gp substrates. Concomitant use of vimseltinib with P-gp substrates may increase exposure of these substrates. (2.3 , 7.1 )
- Breast Cancer Resistance Protein (BCRP) substrates : Avoid concomitant use of ROMVIMZA with BCRP substrates. Concomitant use of vimseltinib with BCRP substrates may increase exposure of these substrates. (7.1 )
- Organic Cation Transporter 2 (OCT) substrates : Avoid concomitant use of ROMVIMZA with OCT2 substrates. Concomitant use of vimseltinib with OCT2 substrates may increase exposure of these substrates. (7.1 )
Effects of ROMVIMZA on Other Drugs
Table 5 describes drug interactions where concomitant use with ROMVIMZA affects another drug.
| P-glycoprotein (P-gp) substrates | |
| Prevention or Management | Avoid concomitant use with P-gp substrates while taking ROMVIMZA. If concomitant use cannot be avoided, take ROMVIMZA at least 4 hours prior to P-gp substrates [ see Dosage and Administration (2.3 ) ] unless otherwise recommended in the substrate Prescribing Information. |
| Mechanism and Clinical Effect(s) | This recommendation is based upon a mechanistic understanding of vimseltinib pharmacokinetics and it being a P-gp inhibitor in vitro [ see Clinical Pharmacology (12.3 ) ]. Concomitant use of ROMVIMZA with P-gp substrates may increase exposure of these substrates; however, this has not been studied clinically. |
| Breast Cancer Resistance Protein (BCRP) substrates | |
| Prevention or Management | Avoid concomitant use with BCRP substrates while taking ROMVIMZA. Refer to the Prescribing Information of the BCRP substrate for dose modifications if concomitant use cannot be avoided. |
| Mechanism and Clinical Effect(s) | This recommendation is based upon a mechanistic understanding of vimseltinib pharmacokinetics and it being a BCRP inhibitor in vitro [ see Clinical Pharmacology (12.3 ) ]. Concomitant use of ROMVIMZA with BCRP substrates may increase exposure of these substrates; however, this has not been studied clinically. |
| Organic Cation Transporter 2 (OCT2) substrates | |
| Prevention or Management | Avoid concomitant use with OCT2 substrates while taking ROMVIMZA. Refer to the Prescribing Information of the OCT2 substrate for dose modifications if concomitant use cannot be avoided. |
| Mechanism and Clinical Effect(s) | This recommendation is based upon a mechanistic understanding of vimseltinib pharmacokinetics and it being an OCT2 inhibitor in vitro [ see Clinical Pharmacology (12.3 ) ]. Concomitant use of ROMVIMZA with OCT2 substrates may increase exposure of these substrates; however, this has not been studied clinically. |
DESCRIPTION
Vimseltinib is a kinase inhibitor. The chemical name of vimseltinib dihydrate is 3-methyl-5-[6-methyl-5-[2-(1-methylpyrazol-4-yl)pyridin-4-yl]oxypyridin-2-yl]-2-(propan-2-ylamino)pyrimidin-4-one, dihydrate.
Vimseltinib is a white to off-white crystalline solid. Vimseltinib is a weak base, very slightly soluble in water. The molecular formula for vimseltinib dihydrate is C 23 H 25 N 7 O 2 • 2 H 2 O, and the molecular weight is 467.52 g/mol. The chemical structure is:
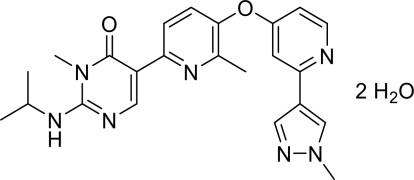
ROMVIMZA (vimseltinib) capsules are supplied as printed hard gelatin capsules containing 14 mg, 20 mg, or 30 mg of vimseltinib (equivalent to 15.18 mg, 21.68 mg, or 32.52 mg of vimseltinib dihydrate, respectively). The capsule contains the following inactive ingredients: crospovidone, lactose monohydrate, and magnesium stearate. The capsule shell contains Brilliant Blue FCF (30 mg strength), erythrosine (30 mg strength), gelatin, Sunset Yellow FCF (14 mg and 20 mg strengths), tartrazine (20 mg), and titanium dioxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Vimseltinib is a kinase inhibitor that inhibits colony-stimulating factor 1 receptor (CSF1R). In vitro, vimseltinib inhibited CSF1R autophosphorylation, signaling induced by CSF1 ligand binding, and proliferation of cells expressing CSF1R.
Pharmacodynamics
Exposure-Response Relationship
Higher vimseltinib exposure is associated with an increased risk of all grades of edema, rash, increased AST, and increased ALT.
Vimseltinib exposure-response relationship for efficacy and time course of pharmacodynamic response have not been fully characterized.
Cardiac Electrophysiology
At the maximum recommended dose of ROMVIMZA, clinically significant QTc interval prolongation was not observed. However, the largest mean increase in QTc interval was 8.2 ms (upper confidence internal = 12.3 ms) after administration of vimseltinib 40 mg once daily for 5 days (3.3 times the maximum recommended weekly dose). The increase in QTc interval was concentration-dependent [ see Clinical Pharmacology (12.3 ) ].
Pharmacokinetics
Vimseltinib pharmacokinetic parameters were determined following a single oral dose of 30 mg or at steady state following multiple doses of 30 mg twice weekly and are provided as mean (CV%) unless otherwise specified.
Vimseltinib peak plasma concentration (C max ) is 283 ng/mL (36%) or 747 ng/mL (39%) after a single dose or at steady state, respectively, and area under the time concentration curve (AUC 0-inf ) is 46,900 ng•h/mL (45%) after a single dose and AUC 0-24hr is 13,400 ng•h/mL (45%) at steady state.
Vimseltinib pharmacokinetics are dose-proportional.
Absorption
Vimseltinib median time to C max (T max ) is 1 hour (0.5 to 4 hours).
Effect of Food
No clinically significant differences in vimseltinib pharmacokinetics were observed following administration of a high-fat meal (800 to 1000 kcal, 50% fat), compared to fasted conditions.
Distribution
Vimseltinib volume of distribution (V/F) is 90 L (16%). Vimseltinib is 96.5% bound to human plasma proteins.
Elimination
Vimseltinib elimination half-life (t 1/2 ) is approximately 6 days (32%) with a clearance (CL/F) of 0.5 L/h (23%).
Metabolism
Vimseltinib is primarily metabolized by oxidation, N -demethylation, and N -dealkylation; secondary biotransformation pathways included N -demethylation, dehydrogenation, and oxidation. CYP450 enzymes are not anticipated to play a major role in the metabolism of vimseltinib.
Excretion
Approximately 43% of the dose was recovered in feces (9.1% unchanged) and 38% in urine (5.1% unchanged) after a single radiolabeled dose.
Specific Populations
No clinically significant differences in the pharmacokinetics of vimseltinib were observed based on age (20 to 91 years), sex, race (Asian, Black or African American, White), body weight (43 to 150 kg), tumor (TGCT or other malignant solid tumors), and mild to moderate renal impairment (estimated glomerular filtration rate [eGFR] ≥30 mL/min calculated by CKD-EPI equation). The effect of severe renal impairment (eGFR <30 mL/min) or moderate to severe hepatic impairment (total bilirubin >1.5 x ULN with any AST) on vimseltinib pharmacokinetics is unknown.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
P-glycoprotein (P-gp) inhibitors:Dabigatran (a P-gp substrate) AUC 0-inf and C max are predicted to increase 2- to 3-fold with concomitant use with vimseltinib 30 mg twice weekly.
Dabigatran C max and AUC 0-inf are predicted to increase up to 1.3-fold if administered 4 hours after administration of vimseltinib 30 mg twice weekly.
Other Drugs:No clinically significant differences in vimseltinib pharmacokinetics were observed when used concomitantly with itraconazole (a P-gp inhibitor) or rabeprazole (a proton pump inhibitor).
In Vitro Studies
CYP 450 enzymes:Vimseltinib is not a substrate of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A.
Vimseltinib is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4.
Vimseltinib is not an inducer of CYP1A2, CYP2B6, or CYP3A4.
Transporter systems:Vimseltinib is a P-gp substrate but is not a substrate of BCRP, BSEP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, and MATE2-K.
Vimseltinib inhibits P-gp, BCRP, BSEP, OATP1B1, OATP1B3, OCT2, BSEP, MATE1, and MATE2-K. Vimseltinib does not inhibit OAT1 and OAT3. Vimseltinib may increase serum creatinine by decreasing renal tubular secretion of creatinine; this may occur due to inhibition of renal transporters OCT2 and MATE1 and may not affect renal function.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 6-month transgenic mouse carcinogenicity study at doses up to 12.5 mg/kg/day, vimseltinib was negative for carcinogenic findings.
In a 2-year rat carcinogenicity study, female rats were orally administered vimseltinib at doses of 0.15, 0.45, or 1.5 mg/kg/day. There was no evidence of vimseltinib-related neoplasms in female rats. Male rats were orally administered vimseltinib at doses of 0.1, 0.3, or 1.0 mg/kg/day. There was a statistically significant increase in the combined incidence of benign and malignant pheochromocytomas in the adrenal gland of male rats administered 1.0 mg/kg/day (approximately 1.4 times the exposure at the recommended dose based on AUC). The relevance of this finding to human carcinogenic risk is not known. In male rats receiving 1.0 mg/kg/day, two out of sixty rats were identified as having sarcomas in the synovium of the femorotibial joint. The sarcoma finding was not statistically significant and its relevance to human carcinogenic risk is not known.
Vimseltinib was not mutagenic in the bacterial reverse mutation assay (Ames). In an in vitro micronucleus assay, vimseltinib increased micronuclei after a 24 -hour incubation in the absence of metabolic activation. In vivo, vimseltinib administered to rats at doses up to 200 mg/kg/day did not increase bone marrow micronucleated polychromatic erythrocytes, nor did vimseltinib increase liver DNA strand breaks.
In a fertility and early embryonic development study, male rats were administered 1, 2.5, or 5 mg/kg/day of vimseltinib starting 10 weeks before cohabitation, during cohabitation with untreated females, and at least 2 weeks post-cohabitation. Lower epididymal and testes weights were observed at 5 mg/kg/day (approximately 12 times the exposure at the recommended dose based on AUC). There were no treatment-related effects on mating, fertility, or sperm parameters at any dose tested. Female rats were administered 2.5, 5, or 10 mg/kg/day of vimseltinib 2 weeks prior to cohabitation with untreated males and during cohabitation until gestational day 7. Although there were no treatment-related effects on mating or estrous cycles, vimseltinib administered daily resulted in post-implantation loss at 10 mg/kg/day in female rats (approximately 20 times the exposure at the recommended dose based on AUC).
In a 26-week repeat-dose general toxicology study, recovery male rats that were administered 2.5 or 5 mg/kg/day had moderate to marked reductions in sperm and marked testicular atrophy (1 of 5 and 2 of 5 animals, respectively) corresponding to approximately 6 and 12 times the exposure at the recommended dose based on AUC, respectively. In a 39-week repeat-dose general toxicology study, minimal to moderate epididymal mineralization occurred in male dogs administered ≥4 mg/kg/day corresponding to exposures lower than the exposure at the recommended dose based on AUC.
Animal Toxicology and/or Pharmacology
In a 26-week repeat-dose general toxicology study in rats, chronic progressive nephropathy occurred at vimseltinib doses of ≥2.5 mg/kg/day (approximately 6 times the exposure at the recommended dose based on AUC). Degeneration of blood vessels (perivascular inflammation and necrosis of arteries and arteriole walls) occurred in multiple tissues at 5 mg/kg/day (approximately 12 times the exposure at the recommended dose based on AUC).
In a 39-week repeat-dose general toxicology study in dogs, skin depigmentation in the head and legs occurred at vimseltinib doses of ≥4 mg/kg/day corresponding to exposures lower than the exposure at the recommended dose based on AUC.
In a 2-year rat carcinogenicity study, synovial hyperplasia and inflammation occurred in the femorotibial joint at vimseltinib doses of ≥0.3 mg/kg/day corresponding to exposures lower than the exposure at the recommended dose based on AUC.
CLINICAL STUDIES
The efficacy of ROMVIMZA was evaluated in MOTION (NCT05059262), a phase 3, double-blind, multicenter, randomized (2:1), placebo-controlled study in patients with TGCT for whom surgical resection may cause worsening functional limitation or severe morbidity. Eligible patients had a confirmed diagnosis of TGCT with measurable disease per the Response Evaluation Criteria in Solid Tumors (RECIST v1.1) with at least one lesion having a minimum size of 2 cm. Patients were randomized to placebo or ROMVIMZA 30 mg twice weekly for 24 weeks. Randomization was stratified by tumor location (lower limb versus all other) and region (United States [US] versus non-US). At Week 25, patients who completed the double-blind, randomized part of the trial were eligible to advance to an ongoing, open-label extension study in which all patients received ROMVIMZA.
The major efficacy outcome measure was overall response rate (ORR) as assessed by blinded independent radiological review (IRR) per RECIST v1.1 at Week 25. Additional efficacy outcomes measured at Week 25 included ORR as assessed using tumor volume score (TVS), mean change from baseline in active range of motion of the affected joint at Week 25 measured by goniometry assessments, change from baseline in the Patient-Reported Outcomes Measurement Information System-Physical Function (PROMIS-PF) 15-item score (upper and lower extremity items), and response of at least a 30% improvement in the mean Brief Pain Inventory (BPI) Worst Pain numeric rating scale (NRS) score without a 30% or greater increase in narcotic analgesic use.
A total of 123 patients were randomized: 83 to ROMVIMZA and 40 to placebo during the double-blind period of the study. The median age was 44 years (range 20 to 78 years); 59% of patients were female; 65% were White, 4% were Asian, 3% were Black or African American, and 28% were not reported or unknown; 69% were not Hispanic or Latino, 3% were Hispanic or Latino, and 28% were not reported or unknown; 74% of patients had prior surgery; 69% of patients had diffuse TGCT; and 23% of patients were previously treated with systemic therapy. Disease locations were knee (67%), ankle (12%), hip (10%), other (5%), foot (3.3%), and wrist (2.4%).
A statistically significant improvement in ORR was demonstrated in patients randomized to ROMVIMZA compared with placebo. Efficacy results in MOTION are summarized in Table 6 .
NR=Not reached; N/A=Not applicable; AMA=American Medical Association; BPI=Brief Pain Inventory; CI=confidence interval; LS=least squares; MMRM=mixed model for repeated measures; n=number of patients in the category; N=sample size; PROMIS-PF=Patient-reported Outcomes Measurement Information System-Physical Function; ROM=range of motion; SD= Standard deviation. | ||
1 DOR results are based on an additional 6 months of follow-up from the time of ORR analysis. | ||
2 The median DOR was estimated using the Kaplan-Meier method. “+” indicates that the patient's response was ongoing at last assessment as of the data cutoff date. | ||
3 Active ROM was normalized to the AMA reference standard. | ||
4 Mean change from baseline was estimated from the MMRM for each corresponding endpoint. Baseline means presented include all patients and not only the ones with data at baseline and Week 25. | ||
5 Data for PROMIS-PF is largely based on lower limb extremity assessment due to tumor location as described above. Higher scores of PROMIS-PF indicate better physical functioning. | ||
6 BPI response in Worst Pain is defined as at least a 30% improvement in the mean BPI Worst Pain NRS score (0-10 NRS) without a 30% or greater increase in narcotic analgesic use at Week 25. | ||
7 95% CI for the difference in response rates based on the stratified Mantel-Haenszel method. | ||
| Efficacy Parameter | ROMVIMZA N = 83 | Placebo N = 40 |
| Overall Response Rate per RECIST v1.1 (95% CI) | 40% (29%, 51%) | 0% (0%, 9%) |
| Complete Response | 5% | 0% |
| Partial Response | 35% | 0% |
| p-value | <0.0001 | |
| Duration of Response 1 | ||
| Median (Range in months) 2 | NR (2.5+, 19.4+) | N/A |
| DOR ≥6 months | 28 (85%) | - |
| DOR ≥9 months | 19 (58%) | - |
| Active ROM 3 | ||
| Patients with data at baseline and Week 25, n | 73 | 33 |
| Baseline mean (SD) | 63.0 (29.4) | 62.9 (32.2) |
| Mean change from baseline 4 (95% CI) | 18.4 (5.6, 31.2) | 3.8 (-10.5, 18.0) |
| Difference in LS means (95% CI) | 14.6% (4.0, 25.3) | |
| p-value | 0.0077 | |
| PROMIS-PF (15-Item score; ranges from 0-100) 5 | ||
| Patients with data at baseline and Week 25, n | 63 | 30 |
| Baseline mean (SD) | 39.0 (6.1 ) | 38.5 (6.0) |
| Mean change from baseline 4 (95% CI) | 4.6 (2.7, 6.5) | 1.3 (-0.5, 3.0) |
| Difference in LS means (95% CI) | 3.3 (1.4, 5.2 ) | |
| p-value | 0.0007 | |
| BPI-30 Response 6 | ||
| Patients with data at baseline and Week 25, n | 68 | 31 |
| Responders, n (%) | 40 (48.2%) | 9 (22.5%) |
| (95% CI) | (37.1%, 59.4%) | (10.8%, 38.5%) |
| Difference in responder rate (95% CI) 7 | 26.2% (9.5, 42.8) | |
| p-value | 0.0056 | |
ORR by TVS was 67% (95% CI: 56, 77) in patients randomized to ROMVIMZA and 0% in patients randomized to placebo; p-value <0.0001.
Individual patient results of change from baseline in active ROM at Week 25 (ROMVIMZA N = 73; placebo N=33) are shown in Figure 1 .
Figure 1: Change from Baseline in Active Range of Motion at Week 25 for MOTION
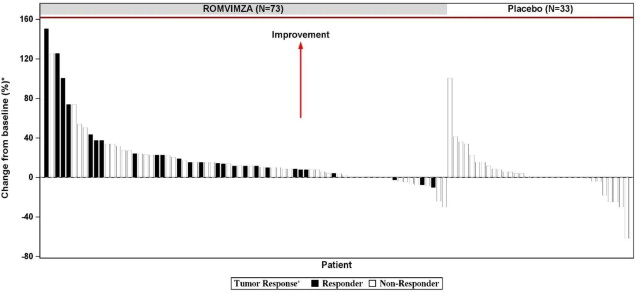
• Percent normal reference range for the affected joint.
+ Response by RECIST v1.1.
HOW SUPPLIED/STORAGE AND HANDLING
| Strength | Description | Package Size and Type | NDC Number |
|---|---|---|---|
| 14 mg | Size 4 hard gelatin capsule with white body and orange cap with black print “DCV14”, packed in oPA-film/ aluminum foil /PVC-film blisters with push-through aluminum foil lidding. | Each carton contains one child-resistant blister pack containing 8 capsules (four-week supply) | 73207-302-40 |
| 20 mg | Size 2 hard gelatin capsule with white body and yellow cap with black print “DCV20”, packed in oPA-film/ aluminum foil /PVC-film blisters with push-through aluminum foil lidding. | Each carton contains one child-resistant blister pack containing 8 capsules (four-week supply) | 73207-303-40 |
| 30 mg | Size 1 hard gelatin capsule with white body and light blue cap with black print “DCV30”, packed in oPA-film/ aluminum foil /PVC-film blisters with push-through aluminum foil lidding. | Each carton contains one child-resistant blister pack containing 8 capsules (four-week supply) | 73207-304-40 |
Store at controlled room temperature 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [See USP Controlled Room Temperature].
Store capsules in their original blister packs until ready to be taken. Do not store ROMVIMZA in another container.
Mechanism of Action
Vimseltinib is a kinase inhibitor that inhibits colony-stimulating factor 1 receptor (CSF1R). In vitro, vimseltinib inhibited CSF1R autophosphorylation, signaling induced by CSF1 ligand binding, and proliferation of cells expressing CSF1R.