Get your patient on Lerochol (Lerodalcibep-Liga)
Lerochol prescribing information
INDICATIONS AND USAGE
LEROCHOL TM is indicated as an adjunct to diet and exercise to reduce low-density lipoprotein cholesterol (LDL-C) in adults with hypercholesterolemia, including heterozygous familial hypercholesterolemia (HeFH).
DOSAGE AND ADMINISTRATION
- The recommended dosage of LEROCHOL is 300 mg administered subcutaneously once monthly. (2.1 )
- Inject LEROCHOL subcutaneously into the abdomen or thigh. A caregiver or healthcare professional can administer into the upper arm. (2.2 )
- Refer to the Instructions for Use for administration of prefilled syringe. (2.2 )
Recommended Dosage
- The recommended dosage of LEROCHOL is 300 mg once monthly administered subcutaneously.
- Assess LDL-C when clinically indicated. The LDL-lowering effect of LEROCHOL may be measured as early as 4 weeks after initiation and, provided monthly dosing is continued, anytime thereafter without regard to timing of the dose.
Recommendations Regarding Missed Dose(s)
- If a dose is missed by:
- Less than 7 days, instruct the patient to administer LEROCHOL as soon as possible and resume the patient's original monthly dosage schedule.
- Seven (7) or more days, instruct the patient to administer LEROCHOL as soon as possible and start a new monthly dosage schedule based on this date.
Important Administration Instructions
- Train patients and/or their caregivers on how to prepare and administer LEROCHOL, according to the Instructions for Use, and instruct them to read and follow the Instructions for Use each time they use LEROCHOL.
- Prior to use, allow LEROCHOL to warm to room temperature up to 25°C (77°F) for at least 30 minutes if LEROCHOL has been refrigerated [see How Supplied/Storage and Handling (16 )] .
- Visually inspect LEROCHOL prior to administration. LEROCHOL is a clear to slightly opalescent, brownish-yellow to amber solution. Do not use if the solution is cloudy or contains particles.
- Inject LEROCHOL subcutaneously into the abdomen or the upper front thighs. If injected by a healthcare professional or caregiver, the back of the upper arms can also be a site of injection [see Instructions for Use] . Do not inject in an area of the skin that is tender, bruised, red, or indurated. Rotate injection sites for each administration.
- To administer the full 300 mg dose, push plunger down until the syringe is empty before removing from the injection site.
DOSAGE FORMS AND STRENGTHS
Injection: 300 mg/1.2 mL (250 mg/mL) of clear to slightly opalescent, brownish-yellow to amber solution in a single-dose prefilled syringe.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Discontinue LEROCHOL when pregnancy is recognized. Alternatively, consider the ongoing therapeutic needs of the individual patient. LEROCHOL increases LDL-C uptake and lowers LDL-C levels in the circulation, thus decreasing cholesterol and possibly other biologically active substances derived from cholesterol; therefore, LEROCHOL may cause fetal harm when administered to pregnant patients based on the mechanism of action [see Clinical Pharmacology (12.1 )]. In addition, treatment of hypercholesterolemia is not generally necessary during pregnancy. Atherosclerosis is a chronic process and the discontinuation of lipid-lowering drugs during pregnancy should have little impact on the outcome of long-term therapy of primary hypercholesterolemia for most patients.
Available data from clinical trials on LEROCHOL use in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes.
In animal reproduction studies, there were no adverse developmental effects observed when pregnant monkeys were administered lerodalcibep-liga subcutaneously during organogenesis and through to parturition at doses up to 100 mg/kg/week [up to 119-fold the exposure at the maximum recommended human dose (MRHD) of 300 mg every month].
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In cynomolgus monkeys, no effects on embryo-fetal or postnatal development (up to 6 months of age) were observed when lerodalcibep-liga was dosed during organogenesis and through parturition at 30 and 100 mg/kg subcutaneously once weekly (exposures up to 119-fold the MRHD by AUC). An assessment of immune function in the infants showed no treatment-related adverse effects. Measurable lerodalcibep-liga serum concentrations were observed in the infant monkeys with infant serum concentrations approximately 2 to 7% of maternal serum concentrations on postnatal days 14, 21 and 28, indicating that lerodalcibep-liga has the potential to be transmitted from the mother to the developing fetus.
Lactation
Risk Summary
There are no data on the presence of lerodalcebip-liga in human milk, the effects on the breastfed infant, or the effects on milk production. In animal reproduction studies, lerodalcibep-liga was present in the milk of lactating monkeys (see Data ). When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for LEROCHOL and any potential adverse effects on the breastfed infant from LEROCHOL or from the underlying maternal condition.
Data
Low concentrations of lerodalcibep-liga were measured in milk samples collected from monkeys on post-partum day (PPD) 7 and PPD 14, when lerodalcibep-liga was dosed during organogenesis through parturition at 30 and 100 mg/kg subcutaneously once weekly (exposures up to 119-fold the MRHD by AUC). There is no information regarding whether lerodalcibep-liga ingested with milk transfers to neonatal or infant monkey circulation.
Pediatric Use
The safety and effectiveness of LEROCHOL in pediatric patients have not been established. Clinical trials of LEROCHOL in pediatric patients with HeFH have not been conducted. Effectiveness of LEROCHOL was not demonstrated in a 24-week, randomized, open-label, active-controlled trial (NCT04034485) in 19 pediatric patients aged 10 to 17 years with homozygous familial hypercholesterolemia (HoFH).
Geriatric Use
Of the 1,631 patients treated with LEROCHOL 300 mg in clinical trials, 687 (42%) patients were 65 years of age and older, while 198 (12%) patients were 75 years of age or older. No overall differences in safety or effectiveness were observed between patients 65 years of age and older and younger adult patients.
Renal Impairment
No dose adjustment is necessary in patients with mild to moderate renal impairment [estimated glomerular filtration rate (eGFR) ≥30 to <90 mL/min] [see Clinical Pharmacology (12.3 )] . LEROCHOL has not been studied in patients with severe renal impairment (eGFR < 30 mL/min) or end-stage renal disease.
Hepatic Impairment
No dose adjustment is necessary in patients with mild (total bilirubin 1.0 to 1.5 upper limit of normal or aspartate aminotransaminase > upper limit of normal) or moderate (total bilirubin 1.5 to 3.0 upper limit of normal) hepatic impairment [see Clinical Pharmacology (12.3 )]. LEROCHOL has not been studied in patients with severe hepatic impairment.
CONTRAINDICATIONS
None.
ADVERSE REACTIONS
Common adverse reactions occurring in ≥1% of patients treated with LEROCHOL were injection site reactions, nasopharyngitis, diarrhea, nausea and peripheral edema. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact LIB Therapeutics, Inc. at 1-877-2-LEROCHOL or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch .
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Adults with Primary Hypercholesterolemia
Adverse Reactions in Two Pooled 52-week Controlled Trials
In two pooled 52-week, double-blind, randomized, placebo-controlled trials (Trials 1 and 2), 1,229 patients received 300 mg of LEROCHOL subcutaneously every 4 weeks [see Clinical Studies (14 )] . The mean age was 64 years (range 25 to 90 years), 52% were 65 years of age or older, 37% female, 79% White, 18% Black or African American, 4% Asian; 7% identified as Hispanic or Latino ethnicity. At baseline, 9% of patients had a diagnosis of HeFH, 74% had established atherosclerotic cardiovascular disease (ASCVD), and 26% were at increased risk for ASCVD. Adverse reactions reported in at least 2% of LEROCHOL-treated patients and more frequently than in placebo-treated patients are shown in Table 1 . Adverse reactions led to treatment discontinuation in 4% of LEROCHOL-treated patients and placebo-treated patients. The most frequent adverse reaction leading to treatment discontinuation was injection site reactions, with a higher frequency in the LEROCHOL-treated group compared to placebo-treated patients (1% vs. 0%).
a Grouped terms composed of several similar terms | ||
| Adverse Reaction a | LEROCHOL 300 mg (N=1,229) % | Placebo (N=612) % |
| Nasopharyngitis | 15 | 14 |
| Injection site reactions | 12 | 5 |
| Peripheral edema | 2 | <1 |
Adverse Reactions in a 24-Week Controlled Trial
In a 24-week, double-blind, randomized, placebo-controlled trial (Trial 3), 318 patients with HeFH received 300 mg of LEROCHOL subcutaneously every 4 weeks [see Clinical Studies (14 )]. Adverse reactions reported in at least 2% of LEROCHOL-treated patients, and more frequently than in placebo-treated patients are shown in Table 2 .
| Adverse Reaction | LEROCHOL 300 mg (N=318) % | Placebo (N=159) % |
|---|---|---|
a Grouped terms composed of several similar terms | ||
| Injection site reactions a | 18 | 3 |
| Nasopharyngitis a | 13 | 9 |
| Diarrhea | 3 | 1 |
| Nausea | 2 | 0 |
| Peripheral edema a | 2 | <1 |
DESCRIPTION
Lerodalcibep-liga is a recombinant fusion protein therapeutic agent comprised of a proprotein convertase subtilisin/kexin type 9 (PCSK9)-binding domain and human serum albumin (HSA). Lerodalcibep-liga is produced in genetically engineered mammalian (Chinese hamster ovary) cells as a single protein with an approximate molecular weight of 77 kDa.
LEROCHOL (lerodalcibep-liga) injection is a clear to slightly opalescent, brownish-yellow to amber, sterile, preservative-free solution for subcutaneous injection. Each single-dose prefilled syringe contains 1.2 mL of lerodalcibep-liga (300 mg), histidine (3.07 mg), L-histidine monohydrochloride (0.88), polysorbate 80 (0.24 mg), sodium chloride (10.52), and Water for Injection. The pH is 6.8.
CLINICAL PHARMACOLOGY
Mechanism of Action
Lerodalcibep-liga is a recombinant fusion protein that binds PCSK9 with picomolar affinity. PCSK9 binds to low-density lipoprotein receptor (LDLR) on the surface of hepatocytes to promote LDLR degradation within the liver. By inhibiting the binding of PCSK9 to LDLR, lerodalcibep-liga increases the number of LDLRs available to clear LDL-C from the blood, thereby lowering LDL-C levels. [see Clinical Pharmacology (12.3 )] .
Pharmacodynamics
After subcutaneous administration of 300 mg lerodalcibep-liga every month, greater than 90% suppression of PCSK9 occurs within 24 hours after dosing and is maintained throughout the dosing interval, returning toward baseline during the last 7 to 10 days of the monthly dosing period.
Pharmacokinetics
Following a single subcutaneous administration, exposure to lerodalcibep-liga increased in a dose proportional manner over the dose range 75 to 300 mg of lerodalcibep-liga.
Lerodalcibep-liga pharmacokinetics were observed at steady state in patients at the approved recommended dosage and are presented as mean (SD), unless otherwise specified. Lerodalcibep-liga maximum concentration (Cmax) is 31.4 (10.2) mcg/mL, and total systemic exposure (AUC0-tau) is 12,600 (5,190) (hrs•mcg/mL) following subcutaneous dose of lerodalcibep-liga 300 mg once every four weeks. Lerodalcibep-liga steady-state is reached following 2 to 3 doses. Lerodalcibep-liga accumulation is approximately 30% at the approved recommended dosage.
Absorption
The median (min, max) estimated subcutaneous bioavailability of lerodalcibep-liga is 83% (66%, 89%). The median (min, max) time to maximum plasma concentration (Tmax) is 6 days (2, 9 days) at steady state.
Distribution
Lerodalcibep-liga does not extensively distribute into tissues; the apparent volume of distribution is 5.3 L.
Elimination
As a protein, lerodalcibep-liga is expected to degrade to small peptides and amino acids. Clearance of free lerodalcibep-liga (CV%) is 0.36 L/day (30%) with an estimated half-life of approximately 10 days. The estimated elimination rate of lerodalcibep-liga (CV%) bound to PCSK9 is 0.47 L/day (22%) which corresponds to a half-life of approximately 1.5 days.
Specific Populations
No clinically significant differences in the pharmacokinetics of LEROCHOL were observed based on age (21 to 78 years), body weight, sex, race, mild (eGFR 60 to 89 mL/min) or moderate (eGFR 30 to 59 mL/min) renal impairment, or mild (total bilirubin 1.0 to 1.5 upper limit of normal or aspartate aminotransaminase greater than the upper limit of normal) or moderate (total bilirubin 1.5 to 3.0 upper limit of normal) hepatic impairment. The effect of severe renal impairment (eGFR less than 30 mL/min) or severe hepatic impairment on LEROCHOL pharmacokinetics is unknown.
Drug Interaction Studies
No formal clinical drug interaction studies have been performed.
Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of lerodalcibep-liga or of other lerodalcibep products.
During the 52-week treatment period in Trial 1 and Trial 2, 15.1% (180/1,195) of LEROCHOL-treated patients at the recommended dosage with evaluable assessments were ADA positive, of whom 22.8% (41/180) had neutralizing antibodies (NAb). During the 24-week treatment period in Trial 3, 11.3% (36/318) of LEROCHOL-treated patients at the recommended dosage with evaluable assessments were ADA positive, of whom 55.6% (20/36) had NAb [see Clinical Studies (14 )] .
There was no identified clinically significant effect of ADA and NAb on the pharmacokinetics, pharmacodynamics (free PCSK9), safety, or effectiveness (LDL-C) of LEROCHOL over the treatment period of 52 weeks.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with lerodalcibep-liga.
The mutagenic potential of lerodalcibep-liga has not been evaluated; however, proteins are not expected to directly interact with DNA or chromosomes.
Fertility studies were not performed. However, in a chronic 6-month toxicology study in sexually mature cynomolgus monkeys, no adverse lerodalcibep-liga-related effects on surrogate markers of fertility (reproductive organ histopathology, menstrual cycling, or sperm parameters) were observed when lerodalcibep-liga was administered subcutaneously at 30 and 100 mg/kg once weekly. The highest dose tested corresponds to 138-fold the recommended human dose of 300 mg every month based on serum AUC.
CLINICAL STUDIES
The efficacy of LEROCHOL was evaluated in three randomized, double-blind, placebo-controlled trials that enrolled 2,017 adults with HeFH, clinical ASCVD, or increased risk for ASCVD, who were on a stable low-fat, low-cholesterol diet and maximally tolerated statin therapy and who required additional LDL-C lowering.
Primary Hypercholesterolemia
Trial 1 (NCT04797247) and Trial 2 (NCT04806893) were both multicenter, double-blind, randomized, placebo-controlled 52-week trials in which 1,844 adults with ASCVD or at increased risk for ASCVD events were randomized 2:1 to receive subcutaneous injections of either LEROCHOL 300 mg (n=1,229) or placebo (n=615) every 4 weeks. Patients were stable on a low-fat, low-cholesterol diet, maximally tolerated dose of statin with or without other oral lipid modifying therapy, and required additional LDL-C reduction.
Baseline Disease and Demographic Characteristics
The mean age in Trial 1 at baseline was 64 years (range: 25 to 90 years), 49% were 65 years or older, 30% were female, 80% were White, 17% were Black or African American, and 3% were Asian; 1.1% identified as Hispanic or Latino ethnicity. Thirty four percent (34%) of patients had diabetes at baseline, 86% established ASCVD, and 14% at increased risk for CVD events. The mean baseline LDL-C was 102 mg/dL. At the time of randomization, 87% of patients were receiving statin therapy and of these 53% were receiving high-intensity statin therapy; 18% were receiving ezetimibe either in combination with a statin or alone.
The mean age in Trial 2 at baseline was 65 years (range: 27 to 87 years), 54% were 65 years or older, 45% were female, 78% were White, 18% were Black or African American, and 4% were Asian; 13% identified as Hispanic or Latino ethnicity. Forty-three percent (43%) of patients had diabetes at baseline, 48% established ASCVD, and 52% at increased risk for CVD events. The mean baseline LDL-C was 116 mg/dL. At the time of randomization, 83% of patients were receiving statin therapy and of these, 39% were on high-intensity doses; 17% were receiving ezetimibe either in combination with a statin or alone.
Endpoint Results
The primary efficacy outcome measure in Trial 1 was the placebo adjusted intent to treat (ITT) analysis of percent change in LDL-C from baseline to Week 52. The difference between the LEROCHOL and placebo group in mean percentage change in LDL-C from baseline to Week 52 was -55% (95% CI: -59.2%, -50.8%; p < 0.0001). For additional results, see Table 3 and Figure 1 .
Abbreviation: ApoB = apolipoprotein B; CI = confidence interval; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol; n = number of patients randomly assigned to each treatment group; LS Mean= least-squares mean | ||||
a LDL-C, Total Cholesterol, non-HDL-C and ApoB at Week 52 were missing for 7.5% and 10.4% of patients assigned to placebo and LEROCHOL, respectively. Multiple imputation washout model was implemented for imputing missing values at Week 52. | ||||
b Least-squares mean from an Analysis of Covariance (ANCOVA) model including treatment as a factor and baseline value as a covariate, assuming unequal variances between groups. | ||||
c p-value <0.001 for superiority, controlled for Type I error rate. | ||||
d Not controlled for Type I error rate. | ||||
| Treatment Group | LDL-C a | Total Cholesterol a | Non-HDL-C a | ApoB a |
| Placebo (n = 308) | -0.1 | -0.2 | -0.7 | +1 |
| LEROCHOL 300mg (n = 614) | -55 | -31 | -46 | -39 |
| Difference from placebo (LS Mean) (95% CI) | -55 b,c (-59, -51) | -31 b,d (-33, -28) | -45 b,d (-48, -41) | -40 b,d (-44, -37) |
Figure 1: Mean Percent Change from Baseline in LDL-C Over 52 Weeks in Patients with Hypercholesterolemia and ASCVD or Increased Risk for ASCVD Events on Maximally Tolerated Statin Therapy (Trial 1)
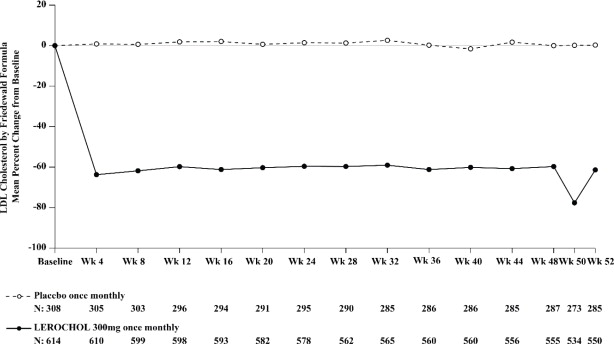
The primary efficacy outcome measure in Trial 2 was the percent change in LDL-C from baseline compared to placebo to Week 52. The difference between the LEROCHOL and placebo groups by ITT analysis in mean percentage change in LDL-C from baseline to Week 52 was -50% (95% CI: -54.2%, -45.2%; p < 0.0001). For additional results, see Table 4 and Figure 2 .
Abbreviation: ApoB = apolipoprotein B; CI = confidence interval; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol; n = number of patients randomly assigned to each treatment group; LS Mean = least-squares mean | ||||
a LDL-C and non-HDL-C at Week 52 were missing for 10.7% and 11.2% of patients assigned to placebo and LEROCHOL, respectively. Total Cholesterol and ApoB at Week 52 were missing for 10.4% and 11.2% of patients assigned to placebo and LEROCHOL, respectively. Multiple imputation washout model was implemented for imputing missing values at Week 52. | ||||
b Least-squares mean from an Analysis of Covariance (ANCOVA) model including treatment and CVD status as factors and baseline value as a covariate, assuming unequal variances between groups. | ||||
c p-value <0.001 for superiority, controlled for Type I error rate. | ||||
d Not controlled for Type I error rate. | ||||
| Treatment Group | LDL-C a | Total Cholesterol a | Non-HDL-C a | ApoB a |
| Placebo (n = 307) | +0.3 | +1 | +1 | +2 |
| LEROCHOL 300mg (n = 615) | -49 | -28 | -41 | -36 |
| Difference from placebo (LS Mean) (95% CI) l | -50 b,c (-54, -45) | -29 b,d (-32, -26) | -42 b,d (-46, -38) | -38 b,d (-42, -35) |
Figure 2: Mean Percent Change from Baseline in LDL-C Over 52 Weeks in Patients with Hypercholesterolemia and ASCVD or Increased Risk for ASCVD Events on Maximally Tolerated Statin Therapy (Trial 2)
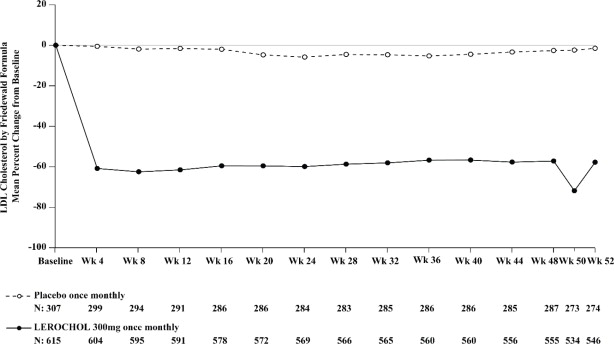
Heterozygous Familial Hypercholesterolemia in Adults
Trial 3 (NCT04797104) was a multicenter, double-blind, randomized, placebo-controlled 24-week trial in which 478 patients with HeFH were randomized 2:1 to receive subcutaneous injections of either LEROHOL 300 mg (n = 319) or placebo (n = 159) every 4 weeks. Patients were stable on diet, maximally tolerated dose of statin, with or without other oral lipid modifying therapy, and required additional LDL-C reduction.
Baseline Disease and Demographic Characteristics
The mean age at baseline was 53 years (range: 18 to 80 years), 19% were aged 65 years or older, 52% were female, 87% were White, 8% were Black or African American, and 5% were Asian; 1% identified as Hispanic or Latino ethnicity. Forty-five percent of patients had diabetes at baseline. The mean baseline LDL-C was 150 mg/dL. At the time of randomization, 89% of patients were receiving statin therapy with 67% receiving high-intensity doses, and 49% also on ezetimibe.
Endpoint Results
The primary efficacy outcome measure in Trial 3 was the percent change in LDL-C from baseline to Week 24. The difference between the LEROCHOL and placebo groups in mean percentage change in LDL-C from baseline to Week 24 was -59% (95% CI: -65.7%, -51.7%; p < 0.0001). For additional results, see Table 5 and Figure 3 .
Abbreviation: ApoB = apolipoprotein B; CI = confidence interval; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol; n = number of patients randomly assigned to each treatment group; LS Mean = least-squares mean | ||||
a LDL-C, Total Cholesterol and non-HDL-C at Week 24 were missing for 3.1% and 2.8% of patients assigned to placebo and LEROCHOL, respectively. ApoB at Week 24 was missing for 3.1% and 3.1% of patients assigned to placebo and LEROCHOL, respectively. Multiple imputation washout model was implemented for imputing missing values at Week 24. | ||||
b Least-squares mean from an Analysis of Covariance (ANCOVA) model including treatment and CVD status as factors and baseline value as a covariate, assuming unequal variances between groups. | ||||
c p-value <0.001 for superiority, controlled for Type I error rate. | ||||
d Not controlled for Type I error rate. | ||||
| Treatment Group | LDL-C a | Total Cholesterol a | Non-HDL-C a | ApoB a |
| Placebo (n = 159) | + 8 | +5 | +7 | +7 |
| LEROCHOL 300mg (n = 319) | -51 | -32 | -44 | -37 |
| Difference from placebo (LS Mean) (95% CI) | -59 b,c (-66, -52) | -37 b,d (-41, -32) | -51 b,d (-57, -45) | -44 b,d (-49, -39) |
Figure 3: Mean Percent Change from Baseline in LDL-C Over 24 Weeks in Patients with HeFH on Maximally Tolerated Statin Therapy (Trial 3)
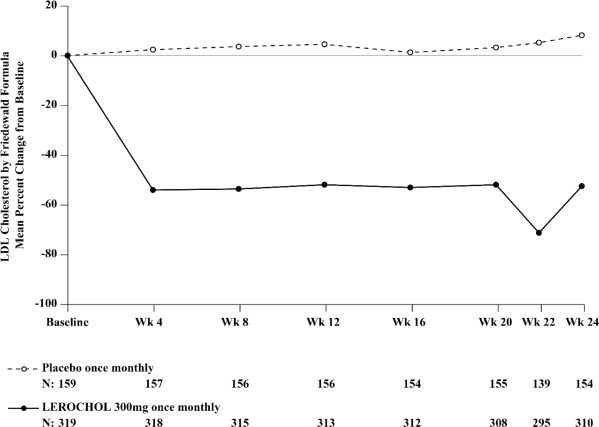
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
LEROCHOL (lerodalcibep-liga) injection 300 mg/1.2 mL (250 mg/mL) is supplied as a single-dose prefilled syringe for subcutaneous injection and is a clear to slightly opalescent, brownish-yellow to amber, sterile, preservative-free solution.
LEROCHOL is supplied in cartons containing one single-dose prefilled syringe (NDC 84685-300-01).
Storage and Handling
Store refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze. Do not shake.
If needed, LEROCHOL may be kept at room temperature up to 25°C (77°F) in the original carton and must be used within 3 months of being removed from the refrigerator. If not used within 3 months, discard LEROCHOL. Do not use beyond the expiration date on the container or package.
INSTRUCTIONS FOR USE
LEROCHOL TM [leer-O-call]
(lerodalcibep-liga)
injection, for subcutaneous use only
Single-dose Prefilled Syringe
This Instructions for Use contains information on how to inject LEROCHOL.
Read and follow these instructions before you start using the LEROCHOL prefilled syringe and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment.
Do not use the LEROCHOL prefilled syringe until your healthcare provider has given you instructions on the right way to give an injection.
Parts of the Prefilled Syringe (seeFigure A )
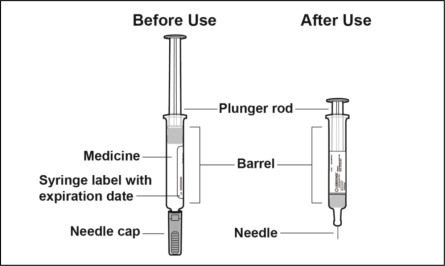
Figure A
Important Information You Need to Know Before Injecting LEROCHOL
- For subcutaneous injection only (inject under the skin).
- Do not use LEROCHOL if the seals on the sides of the carton have already been broken. If seals have already been broken, LEROCHOL may have been tampered with, get a new carton.
- Do not use LEROCHOL if the prefilled syringe is dropped or damaged. Get a new carton.
- Do not re-cap the prefilled syringe as you may stick yourself or damage the needle.
- Do not freeze.
- Safely throw away (dispose of) the used prefilled syringe after the injection is completed (see Step 15. Dispose of used prefilled syringe ).
Preparing to Inject LEROCHOL
Step 1. Gather the supplies needed to give the injection.
- Choose a clean, flat surface in a well-lit area.
- Gather the following supplies and place them on the surface (see Figure B ):
- Carton containing prefilled syringe
Items not included in the carton:
- Alcohol wipe
- Cotton ball or gauze
- Adhesive bandage
- FDA-cleared Sharps disposal container
- Carton containing prefilled syringe
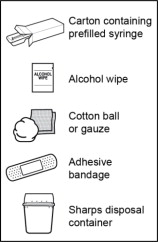
Figure B
Step 2. If refrigerated, let the prefilled syringe warm to room temperature for 30 minutes.
- Let the carton sit at room temperature for 30 minutes before use (see Figure C ).
- Keep the prefilled syringe in the carton during warm-up to protect from light.
- Letting the prefilled syringe warm to room temperature will make the injection more comfortable.
- Do not use a heat source such as a microwave or warm the carton containing the prefilled syringe any other way.
- Do not shake the carton.
Figure C
Step 3. Remove the plastic tray from the carton.
Break the seal and open the side of the carton.
- Do not use LEROCHOL if the seals on the sides of the carton have already been broken. If seals have already been broken, LEROCHOL may have been tampered with, get a new carton.
Gently slide the plastic tray containing the prefilled syringe from the carton into the palm of your hand (Figure D ).
Figure D
Step 4. Remove the prefilled syringe from the plastic tray.
- Place the plastic tray on your hand and peel off the paper seal with your other hand (see Figure E ).
- Turn the plastic tray over and gently press the middle of the tray's back to release the prefilled syringe into the palm of your hand (see Figures F , G and H ).
|
|
|
|
| Figure E | Figure F | Figure G | Figure H |
- Do not pick up or pull the prefilled syringe by the plunger rod or gray needle cap. This could damage the syringe.
- Do not remove the gray needle cap from the prefilled syringe until you are ready to inject.
- Always hold the prefilled syringe by the syringe barrel.
Step 5. Inspect the prefilled syringe.
- Make sure LEROCHOL appears on the prefilled syringe label.
- Check the expiration (EXP) date (see Figure I ).
- Check the prefilled syringe for damage.
- Do not use and get a new carton if LEROCHOL does not appear on the label, the expiration date has passed, or there are signs of damage.
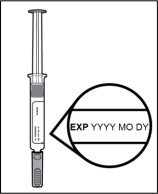
Figure I
Step 6. Inspect the medicine.
Check the medicine to make sure the liquid is clear to brownish-yellow or amber (see Figure J ).
- Do not use the prefilled syringe if the liquid is cloudy or contains visible particles. Get a new carton.
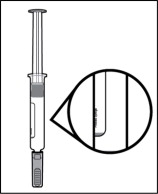
Figure J
Step 7. Wash your hands.
Wash your hands with soap and water and dry them thoroughly (see Figure K ).
Figure K
Step 8. Choose an injection site.
Choose an injection site from the following areas (see Figure L ):
- Stomach area (abdomen) except for the 2-inch (5 cm) area right around your navel (belly button)
- Front of upper legs (thighs)
- Back of upper arms (injection by caregiver or healthcare provider only)
- Do not inject LEROCHOL together with other injectable drugs at the same site.
- Do not select an area of the skin that is tender, bruised, red, or hard. Avoid areas with scars or stretch marks.
- Do not inject through clothing.
- Choose a different site each time you give yourself an injection. If you need to use the same injection site, make sure it is not the same spot on the site you used last time.
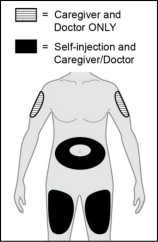
Figure L
Step 9. Clean the injection site.
Clean the skin of the injection site with an alcohol wipe (see Figure M ).
- Do not touch or blow on the injection site again before giving the injection. Let the skin dry before injecting.
Figure M
Injecting LEROCHOL
Step 10. Remove the needle cap.
Hold the barrel of the prefilled syringe with the needle facing away from you.
Firmly grip the needle cap and pull it straight off (see Figure N ).
- Do not twist the needle cap.
- Do not touch the plunger rod as this may cause the prefilled syringe to release medicine prior to injection.
- You may see a drop of liquid at the tip of the needle. This is normal.
- Do not remove any air bubbles in the prefilled syringe.
Dispose of the needle cap right away in an FDA-cleared sharps disposal container (see Figure N ).
- Do not re-cap the prefilled syringe as you may stick yourself or damage the needle.
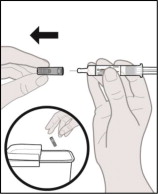
Figure N
Step 11. Pinch the skin and insert the needle.
Hold the barrel of the prefilled syringe in one hand between the thumb and index fingers.
Gently pinch a fold of skin at the injection site with one hand.
With a quick and dart-like motion, insert the needle completely into the pinched skin at a 45 to 90-degree angle (see Figure O ).
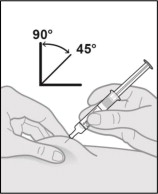
Figure O
Step 12. Give the injection.
Hold the pinch.
Slowly push the plunger rod all the way down until the prefilled syringe is empty (see Figure P ).
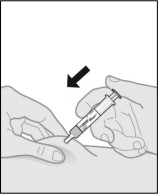
Figure P
Step 13. Remove the prefilled syringe.
- When the prefilled syringe is empty, gently remove the needle from the injection site (see Figure Q ).
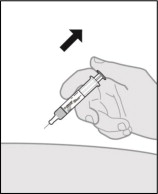
Figure Q
Step 14. Care for the injection site.
If any bleeding happens, gently press a cotton ball or gauze on the injection site and hold for several seconds (see Figure R ). Apply an adhesive bandage if needed.
Figure R
Storing LEROCHOL
- Store LEROCHOL in the refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton until ready to use and to protect from light.
- If needed, LEROCHOL may be kept at room temperature between 68°F to 77°F (20°C to 25°C) in the original carton for up to 3 months after being removed from the refrigerator. Throw away LEROCHOL that has been stored at room temperature for more than 3 months.
- Do not freeze.
- Do not shake.
Keep LEROCHOL and all medicines out of reach of children.
Disposing of LEROCHOL
Step 15. Dispose of used syringe.
Put the used syringe in an FDA-cleared sharps disposal container right away after injection (see Figure S ).
- Do not re-cap the syringe as you may stick yourself.
- Do not reuse the syringe.
- Do not throw away your syringe in your household trash.
- Do not recycle your used FDA-cleared sharps disposal container.
Figure S
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
Disposing of sharps disposal containers:
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your FDA-cleared sharps disposal container.
There may be state or local laws about how you should throw away used needles and syringes.
Additional information about your FDA-cleared sharps disposal container.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at:
http://www.fda.gov/safesharpsdisposal
Manufactured by: LIB Therapeutics, Inc., 5375 Medpace Way, Cincinnati, OH 45227-1543
Distributed by: LIB Therapeutics, Inc., 5375 Medpace Way, Cincinnati, OH 45227-1543
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Approved: December 2025, Version 0.3
Mechanism of Action
Lerodalcibep-liga is a recombinant fusion protein that binds PCSK9 with picomolar affinity. PCSK9 binds to low-density lipoprotein receptor (LDLR) on the surface of hepatocytes to promote LDLR degradation within the liver. By inhibiting the binding of PCSK9 to LDLR, lerodalcibep-liga increases the number of LDLRs available to clear LDL-C from the blood, thereby lowering LDL-C levels. [see Clinical Pharmacology (12.3 )] .