Get your patient on Lazcluze - Lazertinib tablet, Film Coated (Lazertinib)
Lazcluze - Lazertinib tablet, Film Coated prescribing information
INDICATIONS AND USAGE
LAZCLUZE, in combination with amivantamab, is indicated for the first-line treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 L858R substitution mutations, as detected by an FDA-approved test [see Dosage and Administration (2.1) ] .
DOSAGE AND ADMINISTRATION
- The recommended dosage of LAZCLUZE is 240 mg orally once daily with or without food, given in combination with amivantamab. (2.2 )
- Continue treatment until disease progression or unacceptable toxicity. (2.2 )
- Administer LAZCLUZE any time prior to amivantamab when given on the same day. (2.2 )
- Refer to the amivantamab prescribing information for recommended amivantamab dosing information. (2.2 )
- Administer prophylactic and concomitant medications to reduce the risk of dermatologic adverse reactions. (2.3 )
- Administer anticoagulant prophylaxis to reduce the risk of venous thromboembolic events (VTE) for the first four months of treatment. (2.3 )
Patient Selection
Select patients for the first-line treatment of NSCLC with LAZCLUZE, in combination with amivantamab, based on the presence of EGFR exon 19 deletions or exon 21 L858R substitution mutations in tumor or plasma specimens [see Clinical Studies (14) ] . If these mutations are not detected in a plasma specimen, test tumor tissue. Information on FDA-approved tests is available at: http://www.fda.gov/CompanionDiagnostics .
Recommended Dosage and Administration
Recommended Dosage and Administration
The recommended dosage of LAZCLUZE is 240 mg orally once daily administered in combination with amivantamab, with or without food. Swallow LAZCLUZE tablets whole. Do not crush, split, or chew. Continue treatment until disease progression or unacceptable toxicity.
Administer LAZCLUZE any time prior to amivantamab when given on the same day. Refer to the amivantamab prescribing information for recommended amivantamab dosing information.
Missed Dose
If a patient misses a dose of LAZCLUZE within 12 hours, instruct patients to take the missed dose. If more than 12 hours has passed since the dose was to be given, instruct the patient to take the next dose at its scheduled time.
Vomiting
If vomiting occurs any time after taking LAZCLUZE, instruct the patient to take the next dose at its next regularly scheduled time.
Prophylactic and Concomitant Medications
Venous Thromboembolic Events
When initiating treatment with LAZCLUZE in combination with amivantamab, administer anticoagulant prophylaxis to reduce the risk of venous thromboembolic events (VTE) for the first four months of treatment [see Warnings and Precautions (5.1) ]. If there are no signs or symptoms of VTE during the first four months of treatment, consider discontinuation of anticoagulant prophylaxis at the discretion of the healthcare provider.
Dermatologic Adverse Reactions
When initiating treatment with LAZCLUZE in combination with amivantamab, prophylactic and concomitant medications are recommended to reduce the risk and severity of dermatologic adverse reactions [see Warnings and Precautions (5.3) ] .
- Administer an oral antibiotic (doxycycline or minocycline, 100 mg orally twice daily) starting on Day 1 for the first 12 weeks of treatment.
- After completion of oral antibiotic treatment, administer antibiotic lotion to the scalp (clindamycin 1% topical once daily) for the next 9 months of treatment.
- Administer non-comedogenic skin moisturizer (ceramide-based or other formulations that provide long-lasting skin hydration and exclude drying agents) on the face and whole body (except scalp).
- Wash hands and feet with 4% chlorhexidine solution once daily.
- Limit sun exposure during and for 2 months after treatment. Advise patients to wear protective clothing and use broad-spectrum UVA/UVB sunscreen to reduce the risk of dermatologic adverse reactions.
Dosage Modifications for Adverse Reactions
The recommended LAZCLUZE dose reductions for adverse reactions are presented in Table 1.
| Dose at which the adverse reaction occurred | 1 st Dose Reduction | 2 nd Dose Reduction | 3 rd Dose Reduction |
|---|---|---|---|
| 240 mg once daily (one 240 mg tablet) | 160 mg once daily (two 80 mg tablets) | 80 mg once daily (one 80 mg tablet) | Discontinue LAZCLUZE |
The recommended management and dosage modifications of LAZCLUZE for specific adverse reactions are presented in Table 2. Refer to the amivantamab prescribing information for information about dosage modifications for amivantamab.
| Adverse Reaction | Severity | Dosage Modification |
|---|---|---|
| Venous Thromboembolic Events (VTE) [see Warnings and Precautions (5.1) ] | Grade 2 or 3 |
|
| Grade 4 or recurrent Grade 2 or 3 despite therapeutic level anticoagulation |
| |
| Interstitial Lung Disease (ILD)/Pneumonitis [see Warnings and Precautions (5.2) ] | Any Grade |
|
| Dermatologic Adverse Reactions (including dermatitis acneiform, pruritus, dry skin) [see Warnings and Precautions (5.3) ] | Grade 1 |
|
| Grade 2 |
| |
| Grade 3 |
| |
| Grade 4 (including severe bullous, blistering or exfoliating skin conditions) |
| |
| Hepatotoxicity [see Warnings and Precautions (5.4) ] | Grade 3–4 |
|
| Other Adverse Reactions [see Adverse Reactions (6.1) ] | Grade 3–4 |
|
DOSAGE FORMS AND STRENGTHS
Tablets : 80 mg and 240 mg. (3 )
Tablets
- 80 mg tablets: yellow, oval film-coated tablet, debossed with "LZ" on one side and "80" on the other side. Each tablet contains 80 mg of lazertinib.
- 240 mg tablets: reddish purple, oval film-coated tablet, debossed with "LZ" on one side and "240" on the other side. Each tablet contains 240 mg of lazertinib.
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2 )
Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1) ] , LAZCLUZE can cause fetal harm when administered to a pregnant woman. There are no available data on the use of LAZCLUZE in pregnant women to inform a drug-associated risk. Oral administration of lazertinib to pregnant animals during the period of organogenesis resulted in reduced embryo-fetal survival and fetal body weight in rats and malformations in rabbits at exposures approximately 4 and 0.5 times, respectively, the human exposure at the recommended dose of 240 mg/day based on AUC (see Data ) . Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study, pregnant rats received oral doses of 7.5, 30, or 60 mg/kg/day of lazertinib during the period of organogenesis (gestation day 6 to 17). Lazertinib decreased fetal body weights in association with maternal toxicity at 60 mg/kg/day (approximately 4 times the human exposure at the recommended dose of 240 mg/day based on AUC). In a dose range-finding embryo-fetal development study, oral administration of a higher dose of lazertinib (75 mg/kg/day) to pregnant rats during the period of organogenesis resulted in increased post-implantation loss. In an embryo-fetal development study in rabbits, pregnant animals received oral doses of 5, 25, or 45 mg/kg/day of lazertinib during the period of organogenesis (gestation day 7 to 19). Lazertinib caused maternal toxicity (reduced body weight and food consumption leading to moribund condition and early termination) and an increase in the incidence of skeletal malformations in the vertebra and skull (fused maxillary process/zygomatic arch) at 45 mg/kg/day (approximately 0.5 times the human exposure at the recommended dose of 240 mg/day based on AUC).
Lactation
Risk Summary
There are no data on the presence of lazertinib or its metabolites in human milk or their effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with LAZCLUZE and for 3 weeks after the last dose. Refer to the amivantamab prescribing information for lactation information during treatment with amivantamab.
Females and Males of Reproductive Potential
Based on animal data and its mechanism of action, LAZCLUZE can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1) ] .
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating LAZCLUZE.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with LAZCLUZE and for 3 weeks after the last dose. Refer to the amivantamab prescribing information for recommended duration of contraception during treatment with amivantamab.
Males
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with LAZCLUZE and for 3 weeks after the last dose.
Infertility
Based on findings in animals, LAZCLUZE may impair fertility in females and males of reproductive potential. The effects on female fertility were reversible. The effects on male testes in animal studies were not reversible within a 2-week recovery period [see Nonclinical Toxicology (13.1) ].
Pediatric Use
The safety and effectiveness of LAZCLUZE in pediatric patients have not been established.
Geriatric Use
Of the 421 patients with locally advanced or metastatic NSCLC treated with LAZCLUZE in combination with amivantamab in MARIPOSA, 45% were 65 years and older and 12% were 75 years and older. No overall differences in safety or effectiveness were observed between patients aged 65 and older and younger patients.
Renal Impairment
No dose adjustment is recommended in patients with mild or moderate renal impairment (eGFR 30 – 89 mL/min) [see Clinical Pharmacology (12.3) ] .
LAZCLUZE has not been studied in patients with severe renal impairment or end-stage renal disease (eGFR < 30 mL/min).
Hepatic Impairment
No dose adjustment is recommended in patients with mild (total bilirubin ≤ ULN and AST > ULN or total bilirubin ≤ 1.5×ULN and any AST) or moderate (total bilirubin ≤ 1.5 to 3×ULN and any AST) hepatic impairment [see Clinical Pharmacology (12.3) ] .
LAZCLUZE has not been studied in patients with severe hepatic impairment (total bilirubin > 3×ULN and any AST).
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
- Venous Thromboembolic Events (VTE) : Prophylactic anticoagulation is recommended for the first four months of treatment. Monitor for signs and symptoms of VTE and treat as medically appropriate. Withhold LAZCLUZE and amivantamab based on severity. Once anticoagulant treatment has been initiated, resume LAZCLUZE and amivantamab at the same dose at the discretion of the healthcare provider. Permanently discontinue amivantamab and continue LAZCLUZE for recurrent VTE despite therapeutic anticoagulation. (2.3 , 2.4 , 5.1 )
- Interstitial Lung Disease (ILD)/Pneumonitis : Monitor for new or worsening symptoms indicative of ILD/pneumonitis. Withhold LAZCLUZE and amivantamab in patients with suspected ILD/pneumonitis and permanently discontinue if ILD/pneumonitis is confirmed. (2.4 , 5.2 )
- Dermatologic Adverse Reactions : Can cause severe rash including acneiform dermatitis. At treatment initiation, prophylactic and concomitant medications are recommended. Withhold, reduce the dose or permanently discontinue LAZCLUZE and amivantamab based on severity. (2.3 , 2.4 , 5.3 )
- Hepatotoxicity : Can cause severe hepatotoxicity (including increased ALT and AST). Withhold, reduce the dose, or permanently discontinue LAZCLUZE and amivantamab based on severity. (2.4 , 5.4 )
- Ocular Adverse Reactions : Promptly refer patients with new or worsening signs and symptoms of ocular adverse reactions, including keratitis, to an ophthalmologist for evaluation. Withhold, reduce the dose, or permanently discontinue amivantamab and continue LAZCLUZE based on severity. (5.5 )
- Embryo-Fetal Toxicity : Can cause fetal harm. Advise patients of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.6 , 8.1 , 8.3 )
Venous Thromboembolic Events
LAZCLUZE in combination with amivantamab can cause serious and fatal venous thromboembolic events (VTE), including deep venous thrombosis (DVT) and pulmonary embolism (PE). The majority of these events occurred during the first four months of therapy [see Adverse Reactions (6.1) ] .
In MARIPOSA [see Adverse Reactions (6.1) ] , VTE occurred in 36% of patients receiving LAZCLUZE in combination with amivantamab, including Grade 3 in 10% and Grade 4 in 0.5% of patients. On-study VTEs occurred in 1.2% of patients (n=5) while receiving anticoagulation therapy. There were two fatal cases of VTE (0.5%), 7% of patients had VTE leading to dose interruptions of LAZCLUZE, 0.5% of patients had VTE leading to dose reductions of LAZCLUZE, and 1.9% of patients permanently discontinued LAZCLUZE due to VTE. The median time to onset of VTEs was 84 days (range: 6 to 777).
Administer prophylactic anticoagulation for the first four months of treatment [see Dosage and Administration (2.3) ] . The use of Vitamin K antagonists is not recommended. Monitor for signs and symptoms of VTE and treat as medically appropriate.
Withhold LAZCLUZE and amivantamab based on severity [see Dosage and Administration (2.4) ]. Once anticoagulant treatment has been initiated, resume LAZCLUZE and amivantamab at the same dose level at the discretion of the healthcare provider. In the event of VTE recurrence despite therapeutic anticoagulation, permanently discontinue amivantamab. Continue treatment with LAZCLUZE at the same dose level at the discretion of the healthcare provider [see Dosage and Administration (2.4) ] . Refer to the amivantamab prescribing information for recommended amivantamab dosage modification.
Interstitial Lung Disease (ILD)/Pneumonitis
LAZCLUZE in combination with amivantamab can cause interstitial lung disease (ILD)/pneumonitis.
In MARIPOSA [see Adverse Reactions (6.1) ], ILD/pneumonitis occurred in 3.1% of patients treated with LAZCLUZE in combination with amivantamab, including Grade 3 in 1.0% and Grade 4 in 0.2% of patients. There was one fatal case (0.2%) of ILD/pneumonitis and 2.9% of patients permanently discontinued LAZCLUZE and amivantamab due to ILD/pneumonitis [see Adverse Reactions (6.1) ].
Monitor patients for new or worsening symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, fever). Immediately withhold LAZCLUZE and amivantamab in patients with suspected ILD/pneumonitis and permanently discontinue if ILD/pneumonitis is confirmed [see Dosage and Administration (2.4) ].
Dermatologic Adverse Reactions
LAZCLUZE in combination with amivantamab can cause severe rash including dermatitis acneiform, pruritus and dry skin.
In MARIPOSA [see Adverse Reactions (6.1) ] , rash occurred in 86% of patients treated with LAZCLUZE in combination with amivantamab, including Grade 3 in 26% of patients. The median time to onset of rash was 14 days (range: 1 to 556 days). Rash leading to dose reduction of LAZCLUZE occurred in 19% of patients, rash leading to dose interruption of LAZCLUZE occurred in 30% of patients, and LAZCLUZE was permanently discontinued due to rash in 1.7% of patients [see Adverse Reactions (6.1) ].
When initiating treatment with LAZCLUZE in combination with amivantamab, prophylactic and concomitant medications are recommended to reduce the risk and severity of dermatologic adverse reactions [see Dosage and Administration (2.3) ]. Instruct patients to limit sun exposure during and for 2 months after treatment with LAZCLUZE in combination with amivantamab. Advise patients to wear protective clothing and use broad-spectrum UVA/UVB sunscreen.
If skin reactions develop, administer supportive care including topical corticosteroids and topical and/or oral antibiotics. For Grade 3 reactions, administer oral steroids and consider dermatologic consultation. Promptly refer patients presenting with severe rash, atypical appearance or distribution, or lack of improvement within 2 weeks to a dermatologist. Withhold, reduce the dose or permanently discontinue LAZCLUZE and amivantamab based on severity [see Dosage and Administration (2.4) ] .
Hepatotoxicity
LAZCLUZE in combination with amivantamab can cause severe hepatotoxicity (including increased ALT and AST). In MARIPOSA [see Adverse Reactions (6.1) ], based on adverse reaction data, hepatotoxicity occurred in 49% of patients treated with LAZCLUZE, including Grade 3 in 9.3% of patients and Grade 4 in 0.5%.
LAZCLUZE was interrupted for an adverse reaction of hepatotoxicity in 8% of patients, the dose was reduced in 1.4% and permanently discontinued in 0.2%.
Perform liver function tests (including ALT, AST, and total bilirubin) before initiation of LAZCLUZE and during treatment, as clinically indicated. Withhold, reduce the dose, or permanently discontinue LAZCLUZE and amivantamab based on severity [see Dosage and Administration (2.4) ].
Ocular Toxicity
LAZCLUZE, in combination with amivantamab, can cause ocular toxicity, including keratitis.
In MARIPOSA [see Adverse Reactions (6.1) ] , ocular toxicity occurred in 16% of patients treated with LAZCLUZE in combination with amivantamab, including Grade 3 or 4 ocular toxicity in 0.7% of patients. Promptly refer patients presenting with new or worsening eye symptoms to an ophthalmologist. Withhold, reduce the dose or permanently discontinue amivantamab and continue LAZCLUZE based on severity [see Dosage and Administration (2.4) ] .
Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, LAZCLUZE can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lazertinib to pregnant animals during the period of organogenesis resulted in reduced embryo-fetal survival and fetal body weight in rats and malformations in rabbits at exposures approximately 4 and 0.5 times, respectively, the human exposure at the recommended dose of 240 mg/day based on AUC.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with LAZCLUZE and for 3 weeks after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with LAZCLUZE and for 3 weeks after the last dose [see Use in Specific Populations (8.1 , 8.3) ].
ADVERSE REACTIONS
The following adverse reactions are discussed elsewhere in the labeling:
- Venous Thromboembolic Events [see Warnings and Precautions (5.1) ]
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.2) ]
- Dermatologic Adverse Reactions [see Warnings and Precautions (5.3) ]
- Hepatotoxicity [see Warnings and Precautions (5.4) ]
- Ocular Toxicity [see Warnings and Precautions (5.5) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in WARNINGS AND PRECAUTIONS and below reflect exposure to LAZCLUZE in combination with amivantamab in 421 previously untreated patients with locally advanced or metastatic NSCLC whose tumors have EGFR exon 19 deletions or exon 21 L858R substitution mutations in MARIPOSA [see Clinical Studies (14) ] . Patients received LAZCLUZE 240 mg orally once daily in combination with amivantamab intravenously at 1,050 mg (for patients < 80 kg) or 1,400 mg (for patients ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks thereafter starting at week 5. Among the 421 patients who received LAZCLUZE in combination with amivantamab, 84% were exposed to LAZCLUZE for ≥ 6 months and 73% were exposed to LAZCLUZE for > 1 year.
The median age of patients who received LAZCLUZE in combination with amivantamab was 64 years (25 to 88); 64% were female; 59% were Asian, 38% were White, 1.7% were American Indian or Alaska Native, 0.7% were Black or African American, 1% were of unknown or other races; 13% were Hispanic or Latino; 67% had Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 1, 33% had ECOG PS of 0; 60% had EGFR exon 19 deletions, and 40% had EGFR exon 21 L858R substitution mutations.
Serious adverse reactions occurred in 49% of patients who received LAZCLUZE in combination with amivantamab. Serious adverse reactions occurring in ≥ 2% of patients included VTE (11%), pneumonia (4%), rash and ILD/pneumonitis (2.9% each), COVID-19 (2.4%), and pleural effusion and infusion-related reaction (amivantamab) (2.1% each). Fatal adverse reactions occurred in 7% of patients who received LAZCLUZE in combination with amivantamab due to death not otherwise specified (1.2%); sepsis and respiratory failure (1% each); pneumonia, myocardial infarction, and sudden death (0.7% each); cerebral infarction, pulmonary embolism (PE), and COVID-19 infection (0.5% each); and ILD/pneumonitis, acute respiratory distress syndrome (ARDS), and cardiopulmonary arrest (0.2% each).
Permanent discontinuation of LAZCLUZE due to an adverse reaction occurred in 21% of patients. Adverse reactions which resulted in permanent discontinuation of LAZCLUZE in ≥ 1% of patients included ILD/pneumonitis, pneumonia, VTE, rash, respiratory failure, and sudden death.
Dosage interruption of LAZCLUZE due to an adverse reaction occurred in 72% of patients. Adverse reactions which required dosage interruption in ≥ 5% of patients were rash, nail toxicity, COVID-19, VTE, increased ALT, and increased AST.
Dose reductions of LAZCLUZE due to an adverse reaction occurred in 42% of patients. Adverse reactions requiring LAZCLUZE dose reductions in ≥ 5% of patients were rash and nail toxicity.
The most common adverse reactions (≥ 20%) were rash, nail toxicity, infusion-related reaction (amivantamab), musculoskeletal pain, edema, stomatitis, VTE, paresthesia, fatigue, diarrhea, constipation, COVID-19, hemorrhage, dry skin, decreased appetite, pruritus, and nausea. The most common Grade 3 or 4 laboratory abnormalities (≥ 2%) were decreased albumin, decreased sodium, increased ALT, decreased potassium, decreased hemoglobin, increased AST, increased GGT, and increased magnesium.
Table 3 summarizes the adverse reactions (≥ 10%) in MARIPOSA.
| Adverse Reaction | LAZCLUZE in combination with amivantamab (N=421) | Osimertinib (N=428) | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | |
| Skin and subcutaneous tissue disorders | ||||
| Rash Grouped terms | 86 | 26 | 48 | 1.2 |
| Nail toxicity | 71 | 11 | 34 | 0.7 |
| Dry skin | 25 | 1 | 18 | 0.2 |
| Pruritus | 24 | 0.5 | 17 | 0.2 |
| Injury, poisoning and procedural complications | ||||
| Infusion-related reaction Applicable only to amivantamab | 63 | 6 | 0 | 0 |
| Musculoskeletal and connective tissue disorders | ||||
| Musculoskeletal pain | 47 | 2.1 | 39 | 1.9 |
| Gastrointestinal disorders | ||||
| Stomatitis | 43 | 2.4 | 27 | 0.5 |
| Diarrhea | 31 | 2.6 | 45 | 0.9 |
| Constipation | 29 | 0 | 13 | 0 |
| Nausea | 21 | 1.2 | 14 | 0.2 |
| Vomiting | 12 | 0.5 | 5 | 0 |
| Abdominal pain | 11 | 0 | 10 | 0 |
| Hemorrhoids | 10 | 0.2 | 2.1 | 0.2 |
| General disorders and administration site conditions | ||||
| Edema | 43 | 2.6 | 8 | 0 |
| Fatigue | 32 | 3.8 | 20 | 1.9 |
| Pyrexia | 12 | 0 | 9 | 0 |
| Vascular disorders | ||||
| Venous thromboembolism | 36 | 11 | 8 | 2.8 |
| Hemorrhage | 25 | 1 | 13 | 1.2 |
| Nervous system disorders | ||||
| Paresthesia | 35 | 1.7 | 10 | 0.2 |
| Dizziness | 14 | 0 | 10 | 0 |
| Headache | 13 | 0.2 | 13 | 0 |
| Infections and infestations | ||||
| COVID-19 | 26 | 1.7 | 24 | 1.4 |
| Conjunctivitis | 11 | 0.2 | 1.6 | 0 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 24 | 1 | 18 | 1.4 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Cough | 19 | 0 | 23 | 0 |
| Dyspnea | 14 | 1.7 | 17 | 3.5 |
| Eye disorders | ||||
| Ocular toxicity | 16 | 0.7 | 7 | 0 |
| Psychiatric disorders | ||||
| Insomnia | 10 | 0 | 11 | 0 |
Clinically relevant adverse reactions occurring in < 10% of patients who received LAZCLUZE in combination with amivantamab included skin ulcer (applicable to amivantamab) and ILD/pneumonitis.
Table 4 summarizes the laboratory abnormalities in MARIPOSA.
| Laboratory Abnormality | LAZCLUZE in combination with amivantamab (N=421) | Osimertinib (N=428) | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | |
| Chemistry | ||||
| Decreased albumin | 89 | 8 | 22 | 0.2 |
| Increased ALT | 65 | 7 | 29 | 2.6 |
| Increased AST | 52 | 3.8 | 36 | 1.9 |
| Increased alkaline phosphatase | 45 | 0.5 | 15 | 0.5 |
| Decreased calcium (corrected) | 41 | 1.4 | 27 | 0.7 |
| Increased GGT | 39 | 2.6 | 24 | 1.9 |
| Decreased sodium | 38 | 7 | 35 | 5 |
| Decreased potassium | 30 | 5 | 15 | 1.2 |
| Increased creatinine | 26 | 0.7 | 35 | 0.7 |
| Decreased magnesium | 25 | 0.7 | 10 | 0.2 |
| Increased magnesium | 12 | 2.6 | 20 | 4.8 |
| Hematology | ||||
| Decreased platelet count | 52 | 0.7 | 57 | 1.4 |
| Decreased hemoglobin | 47 | 3.8 | 56 | 1.9 |
| Decreased white blood cell | 38 | 1.0 | 66 | 0.7 |
| Decreased neutrophils | 15 | 1.4 | 33 | 1.4 |
DRUG INTERACTIONS
Strong and moderate CYP3A4 inducers: Avoid concomitant use. (7.1 )
Effect of Other Drugs on LAZCLUZE
CYP3A4 Inducers
Avoid concomitant use of LAZCLUZE with strong and moderate CYP3A4 inducers. Consider an alternate concomitant medication with no potential to induce CYP3A4.
Lazertinib is a CYP3A4 substrate. Concomitant use with a strong or moderate CYP3A4 inducer decreased lazertinib concentrations [see Clinical Pharmacology (12.3) ] , which may reduce the efficacy of lazertinib.
Effect of LAZCLUZE on Other Drugs
Certain CYP3A4 Substrates
Monitor for adverse reactions associated with a CYP3A4 substrate where minimal concentration changes may lead to serious adverse reactions, as recommended in the approved product labeling for the CYP3A4 substrate.
Lazertinib is a weak CYP3A4 inhibitor. Concomitant use of LAZCLUZE increased concentrations of CYP3A4 substrates [see Clinical Pharmacology (12.3) ] , which may increase the risk of adverse reactions related to these substrates.
Certain BCRP Substrates
Monitor for adverse reactions associated with a BCRP substrate where minimal concentration changes may lead to serious adverse reactions, as recommended in the approved product labeling for the BCRP substrate.
Lazertinib is a BCRP inhibitor. Concomitant use of LAZCLUZE increased concentrations of BCRP substrates [see Clinical Pharmacology (12.3) ], which may increase the risk of adverse reactions related to these substrates.
DESCRIPTION
LAZCLUZE ® tablets contain lazertinib, a kinase inhibitor for oral use. Lazertinib is present as lazertinib mesylate hydrate with a molecular weight of 668.77 and molecular formula of C 30 H 34 N 8 O 3 ∙CH 4 O 3 S∙H 2 O. The chemical name is N -[5-[[4-[4-[(Dimethylamino)methyl]-3-phenyl-1 H -pyrazol-1-yl]pyrimidin-2-yl]amino]-4-methoxy-2-(morpholin-4-yl)phenyl]acrylamide methanesulfonate hydrate (1:1:1). Lazertinib mesylate hydrate is soluble or practically insoluble in aqueous media, and slightly soluble to freely soluble in organic solvents over a wide range of pH values. The structural formula is:
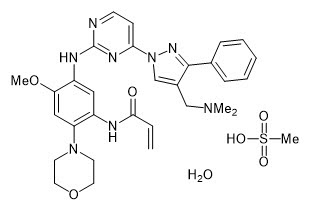
LAZCLUZE (lazertinib) film-coated tablets contain 80 mg or 240 mg of lazertinib, equivalent to 93.86 and 281.58 mg lazertinib mesylate (calculated on anhydrous basis), respectively. The inactive ingredients are croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, and silica hydrophobic colloidal. The tablet coating consists of glycerol monocaprylocaprate type I, iron oxide black (in 240 mg strength tablets), iron oxide red (in 240 mg strength tablets), iron oxide yellow (in 80 mg strength tablets), macrogol (PEG) polyvinyl alcohol graft copolymer, polyvinyl alcohol-partially hydrolyzed, talc, and titanium dioxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Lazertinib is a kinase inhibitor of epidermal growth factor receptor (EGFR) that inhibits EGFR exon 19 deletions and exon 21 L858R substitution mutations at lower concentrations than wild-type EGFR. In human NSCLC cells and mouse xenograft models of EGFR exon 19 deletions or EGFR L858R substitution mutations, lazertinib demonstrated anti-tumor activity. Treatment with lazertinib in combination with amivantamab increased in vivo anti-tumor activity compared to either agent alone in a mouse xenograft model of human NSCLC with an EGFR L858R mutation.
Pharmacodynamics
The exposure-response relationship and time-course of pharmacodynamic response of lazertinib have not been fully characterized.
Cardiac Electrophysiology
At a dose of 320 mg (1.3 times the approved recommended dosage) once daily, a mean increase in the QTc interval > 20 ms is unlikely.
Pharmacokinetics
Lazertinib pharmacokinetics are presented as mean (CV%) for descriptive parameters unless otherwise specified.
Lazertinib maximum plasma concentration (C max ) and area under plasma concentration time curve (AUC) increased dose proportionally from 20 mg to 320 mg (0.08 to 1.3 times the approved recommended dosage) following a single administration and once daily administration. Lazertinib steady state plasma exposure was achieved by day 15 with approximately 2-fold accumulation for AUC.
Absorption
The median time to reach C max is from 2 to 4 hours.
Effect of Food
A high-fat meal (800 to 1000 kcal, approximately 50% fat) did not have a clinically significant effect on lazertinib pharmacokinetics compared to that under fasted conditions.
Distribution
The mean apparent volume of distribution is 2680 L (51%).
Lazertinib is approximately 99.2% bound to human plasma proteins.
Elimination
The mean terminal half-life is 3.7 days (56%).
The mean apparent clearance is 36.4 L/h (47%).
Metabolism
Lazertinib is primarily metabolized by glutathione conjugation, either enzymatic via glutathione-S-transferase (GST) or non-enzymatic, as well as by CYP3A4.
Excretion
Following a single oral dose of radiolabeled lazertinib, approximately 86% of the dose was recovered in feces (< 5% as unchanged) and 4% in urine (< 0.2% as unchanged).
Specific Populations
No clinically significant differences in pharmacokinetics of lazertinib were observed based on age (21 to 88 years), sex, body weight (28 to 122 kg), race (White, Asian, Black or African American), ethnicity (Hispanic/Latino or not Hispanic/Latino), baseline laboratory assessments (creatinine clearance, albumin, alanine aminotransferase, alkaline phosphatase, aspartate aminotransferase), mild or moderate renal impairment (eGFR 30 to 89 mL/min, estimated by CKD-EPI equation), mild [total bilirubin ≤ ULN and AST > ULN or total bilirubin ≤ 1.5 times ULN and any AST] or moderate [total bilirubin ≤ 1.5 to 3×ULN and any AST] hepatic impairment, ECOG performance status, EGFR mutation type, initial diagnosis cancer stage, prior therapies, brain metastasis, and history of smoking.
The effect of severe renal impairment (eGFR 15 to 29 mL/min), end-stage renal disease (eGFR < 15 mL/min) or severe hepatic impairment (total bilirubin > 3 times ULN with any AST) on the pharmacokinetics of lazertinib has not been studied.
GSTM1 Genotype
Patients with at least one GSTM1 normal function allele have 44% lower systemic levels of lazertinib compared with those with the two GSTM1 no-function alleles (i.e., no enzyme activity). No clinically significant differences in safety or efficacy were observed as a function of GSTM1 genotype in patients receiving LAZCLUZE in combination with amivantamab.
Drug Interactions
Clinical Studies and Model-Informed Approaches
Effect of CYP3A4 inducers on lazertinib:
Concomitant use of rifampin (strong CYP3A4 inducer) with LAZCLUZE decreased lazertinib C max by 72% and AUC by 83%.
Concomitant use of efavirenz (moderate CYP3A4 inducer) with LAZCLUZE is predicted to decrease lazertinib steady state C max by at least 32% and AUC by at least 44%.
The effect of concomitant use of weak CYP3A4 inducers on lazertinib C max or AUC is unknown.
Effect of strong CYP3A4 inhibitors on lazertinib:
Concomitant use of itraconazole (strong CYP3A4 inhibitor) with LAZCLUZE increased lazertinib C max by 1.2-fold and AUC by 1.5-fold.
Effect of gastric acid reducing agents on lazertinib:
No clinically significant differences in lazertinib C max and AUC were observed when used concomitantly with gastric acid reducing agents.
Effect of lazertinib on certain CYP3A4 substrates:
Concomitant use of LAZCLUZE increased midazolam (CYP3A4 substrate) C max by 1.4-fold and AUC by 1.5-fold.
Effect of lazertinib on BCRP substrates:
Concomitant use of LAZCLUZE increased rosuvastatin (BCRP substrate) C max by 2.2-fold and AUC by 2-fold.
No clinically significant differences in the pharmacokinetics of the following were observed or predicted when used concomitantly with lazertinib: metformin (OCT1 substrate) or raltegravir (UGT1A1 substrate).
In Vitro Studies
Lazertinib inhibits CYP3A4, UGT1A1, BCRP and OCT1. Lazertinib does not induce CYP1A2, CYP2B6 and CYP3A4.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with lazertinib.
Lazertinib was not genotoxic in an in vitro bacterial reverse mutation (Ames) assay, an in vitro chromosomal aberration assay, or an in vivo micronucleus assay in rats.
In a dedicated fertility and early embryonic development study, male and female rats received oral doses of 7.5, 15, or 30 mg/kg/day of lazertinib. Males were dosed for 10 weeks (29 days prior to pairing, during mating period and continuing post-pairing). Females were dosed for 15 days prior to pairing, during mating period and up to Gestation Day 7. Lazertinib did not have clear effects on estrous cyclicity, mating, fertility, or sperm parameters, but induced an increase in post-implantation loss and decreased number of live fetuses at 30 mg/kg/day (approximately 1.2 times the recommended human dose of 240 mg based on body surface area). In repeat-dose oral general toxicology studies up to 13-weeks duration, lazertinib induced histologic tubular degeneration in the testis; cellular lumen debris, degeneration/necrosis, and reduced sperm in the epididymis; decreased corpora lutea in the ovary; and atrophy in the uterus and vagina. These effects were observed at exposures approximately equivalent to the human exposure at the recommended dose of 240 mg in males, and at exposures approximately 2 times the human exposure at the recommended dose of 240 mg dose in females. Findings in female reproductive organs were reversible. The tubular degeneration in the testis observed in rats at exposures approximately 4 times the human exposure at the recommended dose was not reversible within a 2-week recovery period.
Animal Toxicology and/or Pharmacology
In repeat-dose oral general toxicology studies up to 13-weeks duration in rats and dogs, lazertinib induced multi-organ histologic hyperplasia at exposures approximately equivalent or greater to the human exposure at the recommended dose of 240 mg. Hyperplasia was not reversible in the mandibular lymph node in the 4-week rat study. In rats, lazertinib induced renal toxicity characterized by histologic hyperplasia and inflammation in the kidney at doses ≥ 25 mg/kg (approximately 0.9 times the human exposure at the recommended dose of 240 mg/day based on AUC), along with increased urea nitrogen and histologic papillary necrosis, tubule degeneration/regeneration, and tubule dilatation at exposures approximately 4.4 times the human exposure at the recommended dose of 240 mg/day based on AUC. Increased urea nitrogen, papillary necrosis, and tubule dilatation showed evidence of recovery. In the 13-week toxicology study in dogs, one high dose animal exhibited unilateral tubule cell renal carcinoma at 8 mg/kg/day (approximately 2 times the human exposure at the recommended dose of 240 mg/day based on AUC). Other renal findings in high dose dogs included tubule degeneration/regeneration and infarct, which showed evidence of recovery. In the 4-week toxicology study, lazertinib induced cardiac toxicity in two dogs characterized by histologic findings in the heart (degeneration/necrosis of the myocardium and vessels, fibrosis, hemorrhage, thrombus, mixed cell/vessel inflammation) at 20 mg/kg (approximately 4.8 times the clinical AUC at the 240 mg human dose). One of these dogs also exhibited increased cardiac troponin I and premature ventricular complexes. These cardiac findings were not seen after a 2-week recovery period.
CLINICAL STUDIES
The efficacy of LAZCLUZE, in combination with amivantamab, was evaluated in MARIPOSA [NCT04487080], a randomized, active-controlled, multicenter trial. Eligible patients were required to have untreated locally advanced or metastatic NSCLC with either exon 19 deletions or exon 21 L858R substitution EGFR mutations identified by local testing, not amenable to curative therapy. Patients with asymptomatic or previously treated and stable intracranial metastases were eligible to enroll.
Patients were randomized (2:2:1) to receive LAZCLUZE in combination with amivantamab (N=429), osimertinib monotherapy (N=429), or LAZCLUZE monotherapy (an unapproved regimen for NSCLC) until disease progression or unacceptable toxicity. The evaluation of efficacy for the treatment of untreated metastatic NSCLC relied upon comparison between:
- LAZCLUZE administered at 240 mg orally once daily in combination with amivantamab administered intravenously at 1050 mg (for patients < 80 kg) or 1400 mg (for patients ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks thereafter starting at week 5.
- Osimertinib administered at a dose of 80 mg orally once daily.
Randomization was stratified by EGFR mutation type (exon 19 deletion or exon 21 L858R substitution mutation), Asian race (yes or no), and history of brain metastasis (yes or no). Tumor assessments were performed every 8 weeks for 30 months, and then every 12 weeks until disease progression.
The major efficacy outcome measure was progression-free survival (PFS) as assessed by blinded independent central review (BICR). Additional efficacy outcome measures included overall survival (OS), overall response rate (ORR) and duration of response (DOR).
A total of 858 patients were randomized between the two study arms, 429 to the LAZCLUZE in combination with amivantamab arm and 429 to the osimertinib arm. The median age was 63 (range: 25–88) years; 61% were female; 58% were Asian, and 38% were White, 1.6% were American Indian or Alaska Native, 0.8% were Black or African American, 0.2% were Native Hawaiian or other Pacific Islander, 0.6% were unknown race or multiple races; and 12% were Hispanic or Latino. Eastern Cooperative Oncology Group (ECOG) performance status was 0 (34%) or 1 (66%); 69% never smoked; 41% had prior brain metastases; and 89% had Stage IV cancer at initial diagnosis. Sixty percent of patients had tumors harboring exon 19 deletions and the remaining 40% had exon 21 L858R substitution mutations.
Among the 858 patients with EGFR exon 19 deletion or L858R substitution mutations that were randomized between the amivantamab plus LAZCLUZE arm versus the osimertinib arm, available tissue samples from 544 (63%) patients had evaluable results when tested retrospectively using the cobas EGFR Mutation Test v2. Of the 544 patients with evaluable results, 527 (97%) patients were positive for EGFR exon 19 deletion or L858R substitution mutations, while 17 (3%) patients were negative. Available plasma samples from patients were retrospectively tested using an FDA-approved test to confirm the biomarker status.
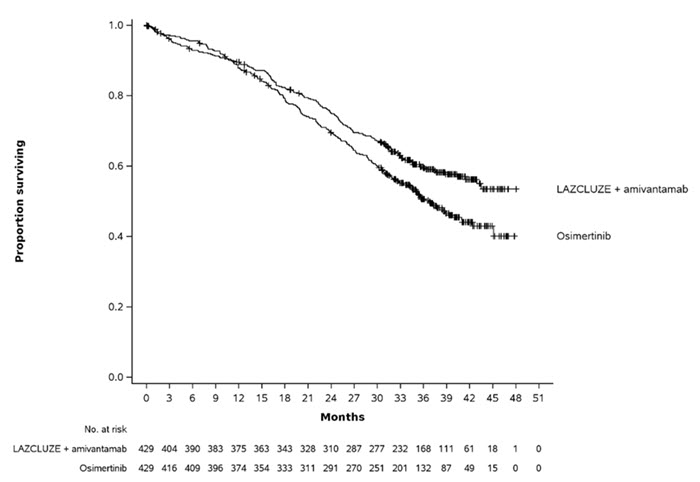
The trial demonstrated a statistically significant improvement in PFS by BICR assessment and OS for LAZCLUZE in combination with amivantamab compared to osimertinib (see Table 5 and Figures 1 and 2 ).
Efficacy results are provided in Table 5.
| LAZCLUZE in combination with amivantamab (N=429) | Osimertinib (N=429) | |
|---|---|---|
| CI = confidence interval; NR = not reached; NE = not estimable | ||
| Progression-free survival (PFS) | ||
| Number of events (%) | 192 (45) | 252 (59) |
| Median, months (95% CI) | 23.7 (19.1, 27.7) | 16.6 (14.8, 18.5) |
| HR Stratified by mutation type (Exon 19del or Exon 21 L858R), prior brain metastases (yes or no), and Asian race (yes or no). , Stratified Cox proportional hazards regression. (95% CI); p-value , Stratified log-rank test. | 0.70 (0.58, 0.85); p=0.0002 | |
| Overall survival (OS) | ||
| Number of events (%) | 173 (40) | 217 (51) |
| Median, months (95% CI) | NR (42.9, NE) | 36.7 (33.4, 41.0) |
| HR , (95% CI); p-value , | 0.75 (0.61, 0.92); p=0.0048 | |
| Overall response rate (ORR) Confirmed responses based on the ITT population. | ||
| ORR, % (95% CI) | 78 (74, 82) | 73 (69, 78) |
| Complete response, % | 5 | 3.5 |
| Partial response, % | 73 | 70 |
| Duration of response (DOR) In confirmed responders. | ||
| Median (95% CI), months | 25.8 (20.1, NE) | 16.7 (14.8, 18.5) |
| Patients with DOR ≥ 6 months Based on observed rates. , % | 86 | 85 |
| Patients with DOR ≥ 12 months, % | 68 | 57 |
Figure 1: Kaplan-Meier Curves of PFS by BICR Assessment in Patients with Previously Untreated NSCLC
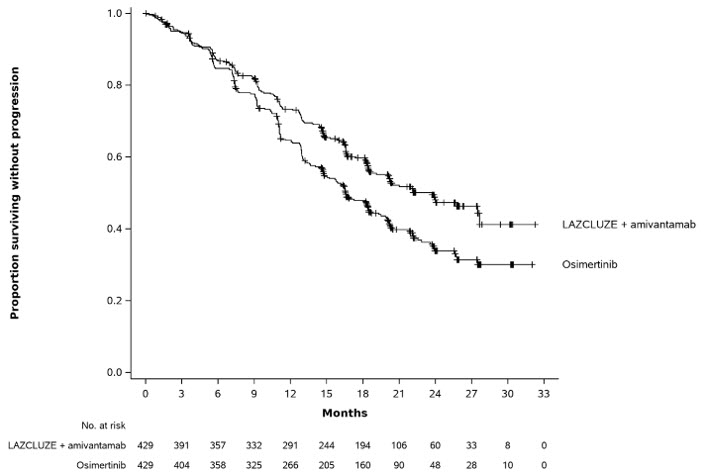
Figure 2: Kaplan-Meier Curves of OS in Patients with Previously Untreated NSCLC
Out of all randomized patients (n=858), 367 (43%) had baseline intracranial lesions assessed by BICR using modified RECIST. Results of pre-specified analyses of intracranial ORR and DOR by BICR in the subset of patients with intracranial lesions at baseline for the LAZCLUZE in combination with amivantamab arm and the osimertinib arm are summarized in Table 6.
| LAZCLUZE in combination with amivantamab (N=180) | Osimertinib (N=187) | |
|---|---|---|
| CI = confidence interval | ||
| Intracranial Tumor Response Assessment | ||
| Intracranial ORR Confirmed responses , % (95% CI) | 68 (60, 75) | 69 (62, 76) |
| Complete response, % | 55 | 52 |
| Intracranial DOR In confirmed responders | ||
| Number of responders | 122 | 129 |
| Patients with DOR ≥ 12 months Based on observed rates , % | 66 | 59 |
| Patients with DOR ≥ 18 months, % | 35 | 23 |
HOW SUPPLIED/STORAGE AND HANDLING
LAZCLUZE ® (lazertinib) tablets are available in the strengths and packages listed below:
| Tablet Strength | Description | Package Configuration | NDC Number |
|---|---|---|---|
| 80 mg | Yellow, oval, film-coated, debossed with "LZ" on one side and "80" on the other side | 60-count bottle | NDC 57894-080-60 |
| 240 mg | Reddish purple, oval, film-coated, debossed with "LZ" on one side and "240" on the other side | 30-count bottle | NDC 57894-240-30 |
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature] .
Mechanism of Action
Lazertinib is a kinase inhibitor of epidermal growth factor receptor (EGFR) that inhibits EGFR exon 19 deletions and exon 21 L858R substitution mutations at lower concentrations than wild-type EGFR. In human NSCLC cells and mouse xenograft models of EGFR exon 19 deletions or EGFR L858R substitution mutations, lazertinib demonstrated anti-tumor activity. Treatment with lazertinib in combination with amivantamab increased in vivo anti-tumor activity compared to either agent alone in a mouse xenograft model of human NSCLC with an EGFR L858R mutation.