Get your patient on Icotyde (Icotrokinra)
Icotyde prescribing information
INDICATIONS AND USAGE
ICOTYDE is indicated for the treatment of moderate-to-severe plaque psoriasis in adults and pediatric patients 12 years of age and older who weigh at least 40 kg who are candidates for systemic therapy or phototherapy.
DOSAGE AND ADMINISTRATION
- See the full prescribing information for recommended evaluation and immunizations prior to treatment. (2.1 )
- Recommended dosage is 200 mg orally once daily. (2.2 )
- Administer ICOTYDE on an empty stomach with water upon waking. (2.2 )
- Wait at least 30 minutes after taking ICOTYDE before eating food. (2.2 )
- For patients who have difficulty swallowing tablets, ICOTYDE can be dispersed in water. (2.3 )
Recommended Evaluation and Immunizations Prior to Treatment Initiation
- Consider evaluating patients for tuberculosis (TB) infection prior to initiating treatment with ICOTYDE based on clinical judgment [see Warnings and Precautions (5.2) ] .
- Complete all age-appropriate vaccinations according to current immunization guidelines [see Warnings and Precautions (5.3) ] .
Recommended Dosage and Administration Instructions
The recommended dosage of ICOTYDE is 200 mg administered orally once daily.
Administer ICOTYDE upon waking on an empty stomach with water. Wait at least 30 minutes after taking ICOTYDE before eating food. Swallow ICOTYDE whole. Do not crush, split, or chew tablets [see Clinical Pharmacology (12.3) ] .
If a patient misses a dose, instruct patients to take the missed dose as soon as possible with a return to normal dosing schedule the following day.
Alternative Preparation and Administration Instructions for Patients Who Have Difficulty Swallowing Tablets
ICOTYDE 200 mg tablet can also be dispersed in water using the following instructions:
- Place one ICOTYDE tablet in a cup containing at least 120 mL (4 ounces) of water. It may take a few minutes for the tablet to disperse. The tablet may not completely disperse. The mixture may look yellow, milky, or cloudy, and small pieces may be seen in the water which are safe to swallow.
- Gently swirl the cup before drinking and swallowing the full mixture.
- Add at least 120 mL (4 ounces) of additional water to the cup and completely swallow the contents to make sure the whole dose is taken.
- Complete administration of ICOTYDE within 15 minutes of dispersion in water.
DOSAGE FORMS AND STRENGTHS
Tablets: 200 mg of icotrokinra, yellowish orange to yellowish brown, oval, film-coated tablets debossed with '200' on one side and 'JNJ' on the other side.
USE IN SPECIFIC POPULATIONS
Moderate or Severe Renal Impairment: Monitor for potential adverse reactions when ICOTYDE is used in patients with eGFR <60 mL/min. (8.6 )
Pregnancy
Risk Summary
The available data on the use of ICOTYDE during pregnancy are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In an animal reproduction study in rabbits, oral administration of icotrokinra to pregnant rabbits during the period of organogenesis at a dose 157 times the maximum recommended human dose (MRHD) based on AUC comparison resulted in maternal body weight loss, low food consumption, late pregnancy loss, and an increased fetal incidence of fused ribs (see Data ) .
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defects, loss, and other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
There is a pregnancy safety study for ICOTYDE. If a patient becomes pregnant while receiving ICOTYDE, healthcare providers can report ICOTYDE exposure by calling 1-800-526-7736 or visiting www.ICOTYDE.com.
Data
Animal Data
In an embryo-fetal development study, icotrokinra was administered to pregnant rats during the period of organogenesis at oral doses of 70, 200, and 1000 mg/kg/day. No maternal or embryo-fetal toxicity was observed at doses up to 1000 mg/kg/day (297 times the MRHD based on AUC comparison). In another embryo-fetal development study, icotrokinra was administered to pregnant rabbits during the period of organogenesis at oral doses of 50, 200, and 500 mg/kg/day. Maternal body weight loss, low food consumption, late pregnancy loss, and an increased fetal incidence of fused ribs were observed at 500 mg/kg/day (157 times the MRHD based on AUC comparison). No maternal or embryo-fetal toxicity was noted at doses up to 200 mg/kg/day in rabbits (27 times the MRHD based on AUC comparison).
In a pre- and post-natal development study in rats, icotrokinra was administered to pregnant rats during pregnancy and lactation periods at oral doses of 20, 70, and 200 mg/kg/day. No maternal or developmental toxicity was noted in doses up to 200 mg/kg/day (127 times the MRHD based on AUC comparison).
Lactation
Risk Summary
There are no data on the presence of icotrokinra in human milk, the effects on the breastfed infant, or the effects on milk production. When administered to lactating rats, icotrokinra was detected in the plasma of nursing pups (see Data ) . When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ICOTYDE and any potential adverse effects on the breastfed infant from ICOTYDE or from the underlying maternal condition.
Data
In a pre- and post-natal development study in rats, icotrokinra was administered orally to pregnant rats during pregnancy and lactation periods at doses up to 200 mg/kg/day (127 times the MRHD based on AUC comparison). Although not directly measured in rat milk, icotrokinra was detected in the plasma of nursing rat pups. No adverse developmental effects were observed in the nursing pups.
Pediatric Use
The safety and effectiveness of ICOTYDE have been established in pediatric patients 12 years of age and older who weigh at least 40 kg with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. Use of ICOTYDE for this indication is supported by evidence from two multi-center, randomized 52-week trials (Trial PSO-3 and Trial PSO-4) conducted in adults and pediatric subjects, including 72 pediatric subjects 12 years of age and older treated with ICOTYDE [see Adverse Reactions (6.1) and Clinical Studies (14) ].
The safety and effectiveness of ICOTYDE in pediatric patients younger than 12 years of age or who weigh less than 40 kg have not been established.
Geriatric Use
Of the 2367 subjects exposed to ICOTYDE in clinical trials for moderate-to-severe plaque psoriasis, 240 (10.1%) were 65 years of age and older, and 36 (1.5%) were 75 years of age and older. No overall differences in safety and effectiveness of ICOTYDE have been observed between subjects 65 years of age and older and younger adult subjects [see Clinical Studies (14) ] .
Renal Impairment
Monitor for potential adverse reactions when ICOTYDE is used in patients with an estimated glomerular filtration rate (eGFR) <60 mL/min [see Clinical Pharmacology (12.3) ] .
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Infections : Avoid treatment with ICOTYDE in patients with any clinically important active infection until the infection resolves or is adequately treated. If such an infection develops, discontinue ICOTYDE until the infection resolves. (5.1 )
Tuberculosis (TB) : Consider evaluating for TB prior to initiating treatment with ICOTYDE based on clinical judgment. Monitor patients for signs and symptoms of active TB during and after treatment with ICOTYDE. (5.2 )
Immunizations : Avoid use of live vaccines during treatment with ICOTYDE. (5.3 )
Infections
Medicines that interact with the immune system may increase the risk of infection.
In the 16-week placebo-controlled trials in subjects with moderate-to-severe plaque psoriasis, the rate of serious infections for ICOTYDE-treated subjects was 0.2% compared to 0.4% of subjects who received placebo.
Avoid treatment with ICOTYDE in patients with any clinically important active infection until the infection resolves or is adequately treated. In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing ICOTYDE. Instruct patients to seek medical advice if signs or symptoms of clinically important infection occur. If a patient develops such an infection and/or is not responding to standard therapy, monitor the patient closely and discontinue ICOTYDE until the infection resolves.
Tuberculosis
Consider evaluating patients for tuberculosis (TB) infection prior to initiating treatment with ICOTYDE based on clinical judgment. Consider anti-TB therapy prior to initiating ICOTYDE in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Monitor patients for signs and symptoms of active TB during and after ICOTYDE treatment. Avoid administering ICOTYDE to patients with active TB.
Immunizations
Avoid use of live vaccines in patients during treatment with ICOTYDE. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Prior to initiating therapy with ICOTYDE, complete immunizations according to current immunization guidelines. No data are available on the response to live or inactive vaccines.
ADVERSE REACTIONS
Most common adverse reactions (≥1%) are headache, nausea, cough, fungal infection, and fatigue. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Biotech, Inc. at 1-800-526-7736 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch .
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ICOTYDE was evaluated in two placebo-controlled trials (Trial PSO-3 and Trial PSO-4) and two placebo- and active-controlled trials (Trial PSO-1 and Trial PSO-2) [see Clinical Studies (14) ] . A total of 2367 adults and pediatric subjects 12 years of age and older who weigh at least 40 kg with moderate-to-severe plaque psoriasis received ICOTYDE 200 mg orally once daily. Of these, 648 subjects were treated with ICOTYDE for at least one year.
Data from these four trials were pooled to evaluate the safety of ICOTYDE compared to placebo for 16 weeks.
Adverse Reactions
Weeks 0 to 16
| Adverse Reactions | ICOTYDE N=1296 n (%) | Placebo N=568 n (%) |
|---|---|---|
| Headache | 51 (4.1) | 19 (3.3) |
| Nausea | 15 (1.2) | 3 (0.5) |
| Cough | 15 (1.2) | 1 (0.2) |
| Fungal Infection Fungal infection includes tinea pedis (n=4), tinea versicolor (n=2), oral candidiasis (n=2), onychomycosis (n=1), skin candida (n=1), urinary tract candidiasis (n=1), vulvovaginal candidiasis (n=1), fungal skin infection (n=1), genital infection fungal (n=1), ear infection fungal (n=1), laryngitis fungal (n=1). Two subjects experienced more than 1 event. | 14 (1.1) | 0 (0) |
| Fatigue | 15 (1.0) | 3 (0.5) |
Adverse reactions that occurred in < 1% of subjects in the ICOTYDE group and at a higher rate than in the placebo group through Week 16 in Trials PSO-1, PSO-2, PSO-3, and PSO-4 were: gastritis, abdominal discomfort, and one fatal case involving upper gastrointestinal bleeding in a subject with underlying risk factors. A relationship of this event to ICOTYDE is not established.
Adverse Reactions in Pediatric Subjects 12 Years of Age and Older
The safety of ICOTYDE was evaluated in pediatric subjects 12 years of age and older who weigh at least 40 kg with moderate-to-severe plaque psoriasis in two placebo-controlled trials (Trial PSO-3 and Trial PSO-4). A total of 72 pediatric subjects were treated with ICOTYDE 200 mg orally once daily. Of these, 45 subjects were treated with ICOTYDE for at least one year. The adverse reactions observed in pediatric subjects were consistent with the most common adverse reactions (≥ 1%) observed in the overall population.
DESCRIPTION
Icotrokinra is an IL-23 receptor antagonist, in the form of a hydrochloride salt. Icotrokinra is a 13-amino acid peptide. The molecular formula for icotrokinra is C 90 H 120 N 20 O 22 S 2 and its molecular weight is 1898.17.
The chemical name for icotrokinra is (4S)-4-[([4-[(2S)-2-[(2S)-2-[([(4R,7S,10S,13S,16S,19R)-19-acetamido-7-(4-acetamidobutyl)-16-(2-amino-2-oxoethyl)-13-[(1R)-1-hydroxyethyl]-3,3,20,20-tetramethyl-10-[(7-methyl-1H-indol-3-yl)methyl]6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentaazacycloicosan-4-yl]carbonyl)amino]-3-[4-(2-aminoethoxy)phenyl]propanamido]-3-(2-naphthyl)propanamido]tetrahydro-2H-pyran-4-yl]carbonyl)amino]-5-[[(2S)-4-amino-1-[[(2S)-1-[(2-amino-2-oxoethyl)(methyl)amino]-1-oxo-3-(pyridin-3-yl)propan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-5-oxopentanoic acid.
The structural formula for icotrokinra hydrochloride is:
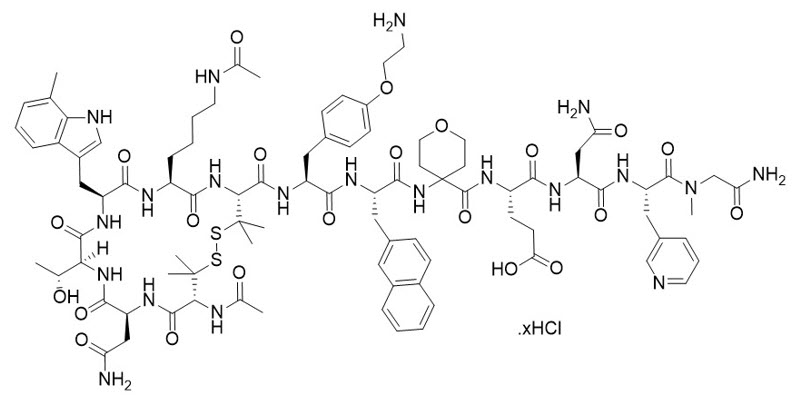
Icotrokinra hydrochloride is a white to almost white powder. It is freely soluble below pH 2, very slightly soluble at pH 5, practically insoluble at pH 9, and freely soluble above pH 11. The isoelectric point (pI) of the compound is 7.15.
ICOTYDE™ (icotrokinra) tablets are supplied as 200 mg film-coated tablets for oral administration. Each tablet of ICOTYDE contains 200 mg icotrokinra (equivalent to 201.6–202.8 mg of icotrokinra hydrochloride) and the following inactive ingredients: colloidal silicon dioxide, crospovidone, magnesium stearate, and silicified microcrystalline cellulose. The film coating contains the following inactive ingredients: glyceryl monocaprylocaprate, iron oxide yellow, macrogol polyvinyl alcohol graft polymer, polyvinyl alcohol partially hydrolyzed, talc, and titanium dioxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Icotrokinra is a peptide that selectively binds to the IL-23 receptor (IL-23R) with a dissociation constant of 7 pM and antagonizes the binding of IL-23. IL-23 is a naturally occurring cytokine that is involved in inflammatory and immune responses. Icotrokinra inhibits the IL-23/IL-23R-dependent release of proinflammatory cytokines.
Pharmacodynamics
Icotrokinra reduced serum levels of proinflammatory cytokines, IL-17A, IL-17F, IL-19, IL-22 and β-defensin-2, relative to pretreatment levels in evaluated subjects with moderate-to-severe plaque psoriasis based on exploratory analysis of the pharmacodynamic markers. A decrease from baseline was observed in the expression of mRNA related to IL-23/Th17 pathway and psoriasis, including molecular targets IL17A , IL17F , IL19 , IL22 , IL23A , and DEFB4A , in lesional skin biopsies up to 24 weeks post treatment in an exploratory analysis of subjects with moderate-to-severe plaque psoriasis. The relationship between these pharmacodynamic markers and the mechanism(s) by which icotrokinra exerts its clinical effects is not fully understood.
Cardiac Electrophysiology
At 5 times the maximum recommended dose of ICOTYDE, clinically significant QTc interval prolongation was not observed.
Pharmacokinetics
Following the administration of icotrokinra 200 mg, the mean (standard deviation) maximum concentration (C max ) is 3.62 (1.48) ng/mL and total systemic exposure (AUC inf ) is 44.8 (11.4) ng•h/mL. Icotrokinra C max and AUC increase in a dose-proportional manner between 0.05 to 5 times the recommended dosage in healthy subjects. Following multiple dose administration accumulation for C max was up to 1.6-fold and for AUC was up to 1.5-fold. Icotrokinra steady state is reached in approximately 3 days.
No clinically relevant differences in icotrokinra pharmacokinetics were observed between healthy subjects and patients with moderate-to-severe plaque psoriasis.
Absorption
Icotrokinra median (min, max) time to maximum plasma concentration (T max ) is 2 (0.25, 8) hours.
Effect of Food
Icotrokinra AUC decreased by 43% and C max decreased by 59% following administration with a high-fat meal (1000 calories, 50% fat) [see Dosage and Administration (2.2) ].
No clinically significant differences in icotrokinra pharmacokinetics were observed following administration of caffeine.
Distribution
Icotrokinra is 52% bound to plasma protein. Blood to plasma partition ratio is 0.53. Icotrokinra steady state apparent (oral) volume of distribution is 92800 L.
Elimination
Icotrokinra median elimination half-life is 12 hours with an apparent (oral) clearance of 6550 L/h.
Metabolism
Icotrokinra is a peptide and it is metabolized by peptide catabolism into smaller peptides.
Excretion
Following oral administration of icotrokinra to healthy subjects, approximately 37% to 81% of the dose was recovered in feces within 24 hours as unchanged icotrokinra, and 0.001% of the dose was recovered in urine as unchanged icotrokinra.
Specific Populations
No clinically significant differences in the pharmacokinetics of icotrokinra were observed based on age (range: 12 to 87 years), body weight (range: 39 to 211 kg), sex, race (76% White, 20.1% Asian, 1.7% Black), ethnicity, immunogenicity, mild (eGFR ≥60 to <90 mL/min, [calculated according to Chronic Kidney Disease Epidemiology Collaboration]) renal impairment. The effect of mild (Child-Pugh Class A) to severe (Child-Pugh Class C) hepatic impairment on icotrokinra pharmacokinetics is unknown. Hepatic impairment is unlikely to affect icotrokinra elimination since the drug is not metabolized hepatically. However, patients with severe hepatic impairment were not studied in clinical trials.
Pediatric Patients
No clinically relevant differences in icotrokinra pharmacokinetics were observed in pediatric patients with moderate-to-severe plaque psoriasis 12 years of age and older who weigh at least 40 kg compared to adults.
Patients with Renal Impairment
Icotrokinra AUC increased by 2.47-fold in patients with moderate (eGFR ≥30 to <60 mL/min, [calculated according to Chronic Kidney Disease Epidemiology Collaboration]) and 2.78-fold in severe (eGFR ≥15 to <30 mL/min) renal impairment. No clinically significant differences in the pharmacokinetics of icotrokinra were observed in patients with mild renal impairment (eGFR ≥60 to <90 mL/min) based on population pharmacokinetic analysis.
Drug Interaction Studies
Clinical Studies
No formal drug-drug interaction studies have been conducted with icotrokinra. No clinically significant drug interactions have been identified.
In Vitro Studies
CYP450 Enzymes: Icotrokinra does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4. Icotrokinra does not induce CYP1A2, CYP2B6, and CYP3A4.
Transporter systems: Icotrokinra is not a substrate of P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, and MATE2-K. Icotrokinra does not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, MATE2-K, and BSEP.
Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of ICOTYDE or of other icotrokinra products.
In four Phase 3 clinical studies through Week 52, 9.6% (199/2083) of subjects treated with ICOTYDE developed anti-drug antibodies. No neutralizing antibodies were detected.
Among ICOTYDE-treated subjects who developed anti-drug antibodies, population pharmacokinetic analysis using pooled data through Week 52 showed that icotrokinra C max increased by 1.2-fold and AUC increased by 1.3-fold.
There was no identified clinically relevant effect of anti-drug antibodies on pharmacokinetics, pharmacodynamics, safety or effectiveness of ICOTYDE over the treatment duration of 52 weeks.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 6-month transgenic rasH2 mouse study, no drug-related tumors were observed at oral doses of icotrokinra up to 500 mg/kg/day (76 times the MRHD based on AUC comparison). In a 2-year rat carcinogenicity study, no drug-related tumors were observed at oral doses of 20 mg/kg/day (16 times the MRHD based on AUC comparison).
Icotrokinra was not genotoxic in an in vitro bacterial reverse mutation assay (the Ames test), an in vitro human lymphocyte chromosomal aberration assay, or an in vivo rat micronucleus and Comet assays.
In male rats, icotrokinra had no adverse effect on mating, fertility or early embryonic development of their offspring at oral doses up to 20 mg/kg/day (114 times the MRHD based on AUC comparison).
In female rats, icotrokinra had no adverse effect on estrous cyclicity, mating, fertility, or early embryonic parameters at oral doses up to 70 mg/kg/day (335 times the MRHD based on AUC comparison).
CLINICAL STUDIES
The efficacy of ICOTYDE was evaluated in four multi-center, randomized, double-blind, placebo and/or active comparator-controlled trials (Trial PSO-1 [NCT06143878], Trial PSO-2 [NCT06220604], Trial PSO-3 [NCT06095115], and Trial PSO-4 [NCT06095102]) that included 2500 subjects (2428 adults and 72 pediatric subjects 12 years and older who weigh at least 40 kg) with moderate-to-severe plaque psoriasis who were eligible for systemic therapy or phototherapy.
Adults with Moderate-to-Severe Plaque Psoriasis (Trials PSO-1 and PSO-2)
Trial Design
Trial PSO-1 and Trial PSO-2 enrolled 1505 adults with moderate-to-severe plaque psoriasis defined as Investigator's Global Assessment (IGA) score ≥3, a Psoriasis Area and Severity Index (PASI) score ≥12, and body surface area (BSA) involvement ≥10%.
Subjects were randomized to either ICOTYDE 200 mg orally once daily, deucravacitinib 6 mg orally once daily or placebo. At Week 16, subjects originally randomized to placebo received ICOTYDE 200 mg orally once daily thereafter. At Week 24, subjects originally randomized to deucravacitinib received ICOTYDE 200 mg orally once daily thereafter.
Baseline Characteristics
Baseline characteristics were consistent across both trials. In Trial PSO-1 and Trial PSO-2, 68% of subjects were male, 78% of subjects were White, 2% of subjects were Black, and 18% of subjects were Asian; for ethnicity, 16% identified as Hispanic or Latino. The mean age was 46 (range: 18 to 86) years, and the mean baseline weight was 88 kg. At baseline, subjects had a median affected BSA of 21%, a median PASI score of 18, and 14% had a history of psoriatic arthritis. The proportion of subjects with a baseline IGA score of 4 (severe) was 20%. At baseline, 72% had received prior systemic therapy, 33% of subjects had received prior phototherapy, and 26% had received prior biologic therapy.
Clinical Response
Trial PSO-1 and Trial PSO-2 assessed responses at Week 16 for ICOTYDE compared to placebo for two co-primary endpoints:
- the proportion of subjects who achieved IGA 0/1 response (defined as IGA score of 0 [cleared] or 1 [minimal] with a ≥2-grade improvement from baseline)
- the proportion of subjects who achieved at least a 90% improvement in PASI scores from baseline (PASI 90).
Other evaluated outcomes for ICOTYDE compared to placebo included IGA 0, PASI 75, PASI 100, Psoriasis Symptoms and Signs Diary (PSSD) Symptom Score of 0, and PSSD Itch Score improvement from baseline (≥4-point reduction).
Other comparisons between ICOTYDE and deucravacitinib that were secondary endpoints included:
- the proportion of subjects who achieved IGA 0/1 score with at least 2-grade improvement from baseline, IGA 0 score, PASI 75, PASI 90, and PASI 100 at Week 16 and Week 24
- the proportion of subjects who achieved PSSD Symptom Score of 0 at Week 16.
Tables 2 and 3 present the efficacy results in adults with moderate-to-severe plaque psoriasis for Trial PSO-1 and Trial PSO-2.
| Endpoint | ICOTYDE (N=311) n (%) | Placebo (N=156) n (%) | Deucravacitinib (N=307) n (%) | Difference, % (95% CI) | |
|---|---|---|---|---|---|
| Difference from Placebo | Difference from Deucravacitinib | ||||
| CI = Confidence interval; PASI = Psoriasis Area and Severity Index; IGA = Investigator's Global Assessment; PSSD = Psoriasis Symptoms and Signs Diary | |||||
| IGA 0 or 1 ("cleared" or "minimal") and a ≥2-grade improvement from baseline) | |||||
| Week 16 Co-primary endpoints comparing ICOTYDE to placebo. | 213 (68) | 17 (11) | 154 (50) | 58 (50, 64) | 18 (11, 26) |
| Week 24 | 230 (74) | - | 161 (52) | - | 22 (14, 29) |
| IGA 0 ("cleared") | |||||
| Week 16 | 114 (37) | 3 (2) | 48 (16) | 35 (29, 41) | 21 (14, 28) |
| Week 24 | 150 (48) | - | 63 (21) | - | 28 (21, 35) |
| PASI 75 | |||||
| Week 16 | 231 (74) | 18 (12) | 176 (57) | 63 (55, 69) | 17 (10, 24) |
| Week 24 | 254 (82) | - | 196 (64) | - | 18 (11, 25) |
| PASI 90 | |||||
| Week 16 | 171 (55) | 6 (4) | 91 (30) | 51 (44, 57) | 25 (18, 33) |
| Week 24 | 205 (66) | - | 127 (41) | - | 25 (17, 32) |
| PASI 100 | |||||
| Week 16 | 97 (31) | 2 (1) | 34 (11) | 30 (24, 36) | 20 (14, 26) |
| Week 24 | 129 (41) | - | 49 (16) | - | 26 (19, 32) |
| PSSD Symptom Score 0 Includes subjects with baseline PSSD Symptom Score >0. | |||||
| N | 286 | 142 | 272 | - | - |
| Week 16 | 68 (24) | 4 (3) | 25 (9) | 21 (15, 27) | 14 (8, 21) |
| PSSD Itch Score Improvement (≥4-point reduction from baseline) Includes subjects with baseline PSSD Itch Score ≥4. | |||||
| N | 251 | 115 | - | - | - |
| Week 4 | 56 (22) | 8 (7) | - | 15 (8, 22) | - |
| Week 16 | 155 (62) | 19 (17) | - | 45 (35, 54) | - |
| Endpoint | ICOTYDE (N=320) n (%) | Placebo (N=81) n (%) | Deucravacitinib (N=322) n (%) | Difference, % (95% CI) | |
|---|---|---|---|---|---|
| Difference from Placebo | Difference from Deucravacitinib | ||||
| CI = Confidence interval; PASI = Psoriasis Area and Severity Index; IGA = Investigator's Global Assessment; PSSD = Psoriasis Symptoms and Signs Diary Trial PSO-2 enrolled 731 subjects, of which 723 subjects were evaluable for efficacy. | |||||
| IGA 0 or 1 ("cleared" or "minimal") and a ≥2-grade improvement from baseline | |||||
| Week 16 Co-primary endpoints comparing ICOTYDE to placebo. | 227 (71) | 7 (9) | 177 (55) | 63 (53, 70) | 16 (9, 23) |
| Week 24 | 220 (69) | - | 179 (56) | - | 13 (6, 21) |
| IGA 0 ("cleared") | |||||
| Week 16 | 118 (37) | 1 (1) | 57 (18) | 36 (29, 42) | 19 (13, 26) |
| Week 24 | 128 (40) | - | 68 (21) | - | 19 (12, 26) |
| PASI 75 | |||||
| Week 16 | 249 (78) | 8 (10) | 198 (61) | 68 (59, 75) | 17 (10, 23) |
| Week 24 | 265 (83) | - | 216 (67) | - | 16 (9, 22) |
| PASI 90 | |||||
| Week 16 | 184 (58) | 1 (1) | 111 (34) | 57 (49, 62) | 23 (16, 30) |
| Week 24 | 208 (65) | - | 141 (44) | - | 21 (14, 29) |
| PASI 100 | |||||
| Week 16 | 102 (32) | 1 (1) | 46 (14) | 31 (24, 36) | 18 (11, 24) |
| Week 24 | 107 (33) | - | 52 (16) | - | 17 (11, 24) |
| PSSD Symptom Score 0 Includes subjects with baseline PSSD Symptom Score >0. | |||||
| N | 296 | 70 | 282 | - | - |
| Week 16 | 64 (22) | 0 | 36 (13) | 22 (15, 27) | 9 (3, 15) |
| PSSD Itch Score Improvement (≥4-point reduction from baseline) Includes subjects with baseline PSSD Itch Score ≥4. | |||||
| N | 256 | 60 | - | - | - |
| Week 4 | 54 (21) | 3 (5) | - | 16 (6, 22) | - |
| Week 16 | 154 (60) | 9 (15) | - | 46 (34, 56) | - |
Adults and Pediatric Subjects 12 Years of Age and Older with Moderate-to-Severe Plaque Psoriasis (Trial PSO-3)
Trial Design
Trial PSO-3 enrolled 684 subjects (618 adults and 66 pediatric subjects 12 years of age and older who weigh at least 40 kg) with moderate-to-severe plaque psoriasis defined as Investigator's Global Assessment (IGA) score ≥3, a Psoriasis Area and Severity Index (PASI) score ≥12, and BSA ≥10%.
Subjects were randomized to receive either ICOTYDE (200 mg orally once daily) or placebo for 16 weeks.
Baseline Characteristics
In Trial PSO-3, 65% of subjects were male, 72% of the subjects were White, 1% of the subjects were Black, and 24% of subjects were Asian; for ethnicity, 13% identified as Hispanic or Latino. The mean age was 43 (range: 12 to 85) years, the mean baseline weight was 86 kg, and 10% were 12 years to less than 18 years of age. At baseline, subjects had a median affected BSA of 20%, a median PASI score of 17, and 13% had a history of psoriatic arthritis. The proportion of subjects with a baseline IGA score of 4 (severe) was 25%. At baseline, 72% had prior systemic treatment, 30% of subjects had received prior phototherapy, and 34% had received prior biologic therapy.
Clinical Response
Trial PSO-3 assessed responses at Week 16 for ICOTYDE compared to placebo for two co-primary endpoints:
- the proportion of subjects who achieved IGA 0/1 response (defined as IGA score of 0 [cleared] or 1 [minimal] with a ≥2-grade improvement from baseline)
- the proportion of subjects who achieved PASI 90.
Other evaluated outcomes for ICOTYDE compared to placebo included IGA 0, PASI 75, PASI 100, PSSD Symptom Score of 0, and PSSD Itch Score improvement from baseline (≥4-point reduction).
Table 4 presents the efficacy results in adults and pediatric subjects 12 years of age and older for Trial PSO-3.
| Endpoint | ICOTYDE (N=456) n (%) | Placebo (N=228) n (%) | Difference, % from Placebo (95% CI) |
|---|---|---|---|
| CI = Confidence interval; PASI = Psoriasis Area and Severity Index; IGA = Investigator's Global Assessment; PSSD = Psoriasis Symptoms and Signs Diary | |||
| IGA 0 or 1 ("cleared" or "minimal") and a ≥2-grade improvement from baseline | |||
| Week 16 Co-primary endpoints comparing ICOTYDE to placebo. | 295 (65) | 19 (8) | 56 (50, 62) |
| IGA 0 ("cleared") | |||
| Week 16 | 152 (33) | 3 (1) | 32 (27, 37) |
| PASI 75 | |||
| Week 16 | 315 (69) | 25 (11) | 58 (52, 64) |
| PASI 90 | |||
| Week 16 | 226 (50) | 10 (4) | 45 (40, 50) |
| PASI 100 | |||
| Week 16 | 123 (27) | 1 (<1) | 26 (22, 31) |
| PSSD Symptom Score 0 Includes subjects with baseline PSSD Symptom Score >0. | |||
| N | 408 | 208 | - |
| Week 16 | 82 (20) | 2 (<1) | 19 (15, 24) |
| PSSD Itch Score Improvement (≥4-point reduction from baseline) Includes subjects with baseline PSSD Itch Score ≥4. | |||
| N | 350 | 176 | - |
| Week 4 | 67 (19) | 9 (5) | 14 (9, 19) |
| Week 16 | 203 (58) | 23 (13) | 45 (37, 52) |
Maintenance and Durability of Clinical Response
In Trial PSO-3, adults who were randomized to ICOTYDE 200 mg orally once daily and were PASI 75 responders or IGA 0 or 1 responders at Week 24 were re-randomized to continue ICOTYDE 200 mg orally once daily or withdrawn from therapy (i.e., received placebo).
For adults who were re-randomized and had a PASI 90 response at Week 24, 84% (108/128) of subjects who continued on ICOTYDE maintained PASI 90 response at Week 52 compared to 21% (27/129) of subjects randomized to placebo. For PASI 90 responders at Week 24 who were re-randomized to placebo, the median time to loss of PASI 90 response was approximately 10 weeks.
For adults who were re-randomized and had an IGA 0/1 response at Week 24, 82% (123/150) of subjects who continued on ICOTYDE maintained IGA 0/1 response at Week 52 compared to 23% (35/150) of subjects randomized to placebo. For IGA 0/1 responders at Week 24 who were re-randomized to placebo, the median time to loss of IGA 0/1 response was approximately 10 weeks.
Pediatric Subjects 12 Years of Age and Older with Moderate-to-Severe Plaque Psoriasis
Trial PSO-3 included 66 pediatric subjects 12 years of age and older who weigh at least 40 kg.
Table 5 presents the efficacy results in pediatric subjects 12 years of age and older at Week 16 enrolled in Trial PSO-3.
| Endpoint | ICOTYDE n (%) | Placebo n (%) | Difference, % from Placebo (95% CI) |
|---|---|---|---|
| CI = Confidence interval; PASI = Psoriasis Area and Severity Index; IGA = Investigator's Global Assessment | |||
| Number of randomized pediatric subjects | 44 | 22 | - |
| IGA 0 or 1 ("cleared" or "minimal") and a ≥2-grade improvement from baseline | 37 (84) | 6 (27) | 56 (33, 74) |
| PASI 90 | 31 (70) | 3 (14) | 56 (33, 73) |
Adults and Pediatric Subjects 12 Years of Age and Older with Moderate-to-Severe Plaque Psoriasis of the Scalp or Genital Area (Trial PSO-4)
Trial Design
Trial PSO-4 enrolled 311 subjects (305 adults and 6 pediatric subjects 12 years of age and older who weigh at least 40 kg) with moderate-to-severe plaque psoriasis who had a minimum BSA involvement of ≥1%, an IGA score of ≥2 and had failed to respond to at least one topical therapy for the treatment of plaque psoriasis. Additionally, subjects in Trial PSO-4 had at least one of the following baseline conditions: ss-IGA score ≥3 (at least moderate plaque psoriasis of the scalp), static Physician's Global Assessment of Genitalia (sPGA-G) score ≥3 (at least moderate plaque psoriasis of the genital area), and/or Physician's Global Assessment of Hands and/or Feet (hf-PGA) score ≥3 (at least moderate plaque psoriasis of the hands and/or feet).
Subjects were randomized to receive either ICOTYDE 200 mg orally once daily or placebo for 16 weeks. At Week 16, subjects originally randomized to placebo switched to receive ICOTYDE 200 mg orally once daily, and subjects randomized to ICOTYDE at baseline remained on treatment through the end of study.
Baseline Characteristics
In Trial PSO-4, 64% of subjects were male, 78% of subjects were White, 1% of subjects were Black, and 20% of subjects were Asian; for ethnicity, 7% identified as Hispanic or Latino. The mean age was 45 (range: 12 to 87) years, the mean baseline weight was 86 kg, and 2% were 12 years to less than 18 years of age. The proportion of subjects with affected BSA less than 10% was 36% with a median affected BSA of 12%. Subjects had a median PASI score of 14, and 16% had a history of psoriatic arthritis. The proportion of subjects with a baseline IGA score of 3 (moderate), and 4 (severe) were 73% and 22%, respectively. The proportion of subjects with ss-IGA score of 3 or greater was 81%. The proportion of subjects with sPGA-G score of 3 or greater was 45%. The proportion of subjects with hf-PGA score of 3 or greater was 23%. Scalp, genital and hand/foot subpopulations were not mutually exclusive. At baseline, 73% had received prior systemic therapy, 39% of subjects had received prior phototherapy, and 33% had received prior biologic therapy.
Clinical Response
In Trial PSO-4, the primary endpoint was the proportion of subjects who achieved an IGA 0/1 response (defined as IGA score of 0 [cleared] or 1 [minimal] and a ≥2-grade improvement from baseline at Week 16).
Other secondary endpoints at Week 16 included proportion of subjects who achieved ss-IGA score of 0 (absence of disease) or 1 (very mild disease), Psoriasis Scalp Severity Index (PSSI) 90, improvement in scalp itch as measured by Scalp Itch Numerical Rating Scale (NRS) Score, sPGA-G score of 0 (clear) or 1 (minimal), improvement of genital itch severity as measured by a reduction of at least 4 points in the 11-point Genital Psoriasis Symptoms Scale (GPSS) Genital Itch NRS Score, and the patient-perceived impact of psoriasis of the genital area on limiting frequency of sexual activity (intercourse or other activities) as measured by the Genital Psoriasis Sexual Frequency Questionnaire (GenPs-SFQ) Item 2.
Table 6 presents the efficacy results in adults and pediatric subjects 12 years of age and older at Week 16 for Trial PSO-4.
| Endpoint | ICOTYDE n (%) | Placebo n (%) | Difference, % from Placebo (95% CI) |
|---|---|---|---|
| CI = Confidence interval; IGA = Investigator's Global Assessment; ss-IGA = Scalp-specific Investigator Global Assessment; PSSI = Psoriasis Scalp Severity Index; NRS = Numerical Rating Scale; sPGA-G = Static Physician's Global Assessment of Genitalia; GPSS = Genital Psoriasis Symptoms Scale; GenPs-SFQ = Genital Psoriasis Sexual Frequency Questionnaire | |||
| Number of randomized subjects | 208 | 103 | - |
| IGA 0 or 1 ("cleared" or "minimal") and a ≥2-grade improvement from baseline Primary endpoint comparing ICOTYDE to placebo. | 118 (57) | 6 (6) | 51 (42, 59) |
| Number of subjects with baseline ss-IGA score of ≥3 | 167 | 85 | - |
| ss-IGA 0 or 1 ("absence of disease" or "very mild disease") (scalp) | 110 (66) | 9 (11) | 56 (45, 64) |
| PSSI 90 | 96 (57) | 5 (6) | 52 (42, 60) |
| Number of subjects with baseline Scalp Itch NRS score ≥4 and baseline ss-IGA score ≥3 | 131 | 58 | - |
| Scalp Itch NRS score (≥4-point improvement) | 77 (59) | 5 (9) | 50 (38, 61) |
| Number of subjects with baseline sPGA-G score of ≥3 | 98 | 42 | - |
| sPGA-G 0 or 1 ("clear" or "minimal") (genitalia) | 75 (77) | 9 (21) | 55 (39, 68) |
| Number of subjects with baseline GPSS Genital Itch NRS score ≥4 and baseline sPGA-G score ≥3 | 69 | 31 | - |
| GPSS Genital Itch NRS score (≥4-point improvement) | 44 (64) | 4 (13) | 50 (31, 64) |
| Number of subjects with baseline GenPs-SFQ Item 2 score ≥2 and baseline sPGA-G score ≥3 | 55 | 25 | - |
| GenPs-SFQ Item 2 score 0 or 1 ("never" or "rarely") (genitalia) | 44 (80) | 9 (36) | 43 (20, 62) |
Subgroup Analyses of Trials PSO-1, PSO-2, PSO-3 and PSO-4
An examination of age, gender, race, body weight, baseline disease severity, and previous treatment with systemic or biologic agents did not identify differences in response to ICOTYDE among these subgroups.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ICOTYDE™ (icotrokinra) tablets: 200 mg, yellowish orange to yellowish brown, oval, film-coated tablets debossed with '200' on one side and 'JNJ' on the other side.
ICOTYDE is supplied in bottles of 30 tablets (NDC Number: 57894-201-30), with a silica gel desiccant and a child-resistant closure.
Storage and Handling
Store at 20 °C to 25 °C (68 °F to 77 °F); excursions permitted between 15 °C to 30 °C (59 °F to 86 °F) [see USP Controlled Room Temperature]. Store in original package to protect from moisture. Do not discard desiccant.
Mechanism of Action
Icotrokinra is a peptide that selectively binds to the IL-23 receptor (IL-23R) with a dissociation constant of 7 pM and antagonizes the binding of IL-23. IL-23 is a naturally occurring cytokine that is involved in inflammatory and immune responses. Icotrokinra inhibits the IL-23/IL-23R-dependent release of proinflammatory cytokines.