Get your patient on Hernexeos - Zongertinib tablet, Film Coated (Zongertinib)
Hernexeos - Zongertinib tablet, Film Coated prescribing information
| Indications and Usage (1 ) | 2/2026 |
INDICATIONS AND USAGE
HERNEXEOS is indicated for the treatment of adult patients with unresectable or metastatic non-squamous non-small cell lung cancer (NSCLC) whose tumors have HER2 (ERBB2) tyrosine kinase domain activating mutations, as detected by an FDA-authorized test [see Dosage and Administration (2.1) ] .
This indication is approved under accelerated approval based on objective response rate and duration of response [see Clinical Studies (14) ] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
DOSAGE AND ADMINISTRATION
- Select patients for treatment with HERNEXEOS based on the presence of HER2 (ERBB2) tyrosine kinase domain activating mutations. (2.1 )
- The recommended dosage of HERNEXEOS is based on body weight: (2.2 )
- Take HERNEXEOS orally once daily with or without food until disease progression or unacceptable toxicity. (2.2 )
Patient Selection
Select patients for treatment of unresectable or metastatic NSCLC based on the presence of HER2 (ERBB2) tyrosine kinase domain activating mutations in tumor specimens [see Clinical Studies (14) ] . Information on FDA-authorized tests for HER2 (ERBB2) tyrosine kinase domain activating mutations is available at: http://www.fda.gov/CompanionDiagnostics.
Recommended Dosage and Administration
The recommended dosage of HERNEXEOS is based on body weight:
- Patients weighing less than 90 kg: 120 mg
- Patients weighing 90 kg or greater: 180 mg
Take HERNEXEOS orally once daily with or without food until disease progression or unacceptable toxicity. Swallow HERNEXEOS tablets whole with water. Do not split, crush, or chew tablets.
Missed Dose
If a dose is missed within 12 hours, take the dose. If a dose is missed by more than 12 hours, skip the missed dose and take the next scheduled dose.
Vomited Dose
If a dose is vomited, do not take an additional dose. Take the next dose at the regularly scheduled time.
Dosage Modifications for Adverse Reactions
The recommended dose reductions for adverse reactions are presented in Table 1.
| Current HERNEXEOS Dose | First Reduction | Second Reduction |
|---|---|---|
| 180 mg | 120 mg | 60 mg |
| 120 mg | 60 mg | Permanently discontinue |
| Permanently discontinue HERNEXEOS in patients who are unable to tolerate 60 mg once daily. | ||
The recommended dosage modifications for adverse reactions are presented in Table 2.
| Adverse Reaction | Severity• | Dosage Modification |
|---|---|---|
| ALT = alanine aminotransferase; AST = aspartate aminotransferase; ULN = upper limit of normal | ||
| •Based on Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 | ||
| Hepatotoxicity [see Warnings and Precautions (5.1) ] | Grade 3 or 4 ALT and/or AST withoutincreased total bilirubin |
|
| Grade 3 total bilirubin |
| |
| Grade 4 total bilirubin |
| |
| ALT or AST ≥ 3× ULN withtotal bilirubin ≥ 2× ULN |
| |
| Left Ventricular Dysfunction [see Warnings and Precautions (5.2) ] | LVEF 40 to 50% and decrease from baseline of 10 to 19% |
|
| LVEF 20 to 39% or ≥ 20% decrease from baseline |
| |
| Symptomatic Congestive Heart Failure |
| |
| Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.3) ] | Grade 2 |
|
| Grade 3 or Grade 4 |
| |
| Diarrhea [see Adverse Reactions (6.1) ] | Grade 2 |
|
| Grade 2 lasting ≥ 2 days despite anti-diarrheal treatment |
| |
| Grade 3 or Grade 4 |
| |
| Other Adverse Reactions [see Adverse Reactions (6.1) ] | Grade 3 |
|
| Grade 4 |
| |
Dosage Modifications for Drug Interactions
Strong CYP3A Inducers
Avoid concomitant use of strong CYP3A inducers with HERNEXEOS .
If concomitant use cannot be avoided, increase the HERNEXEOS dose based on body weight [see Drug Interactions (7.1) ] :
- Patients weighing less than 90 kg: from 120 mg to 240 mg
- Patients weighing 90 kg or greater: from 180 mg to 360 mg
After discontinuing a CYP3A inducer, resume the HERNEXEOS dose (7 to 14 days after discontinuing the CYP3A inducer) that was taken prior to initiating the CYP3A inducer.
DOSAGE FORMS AND STRENGTHS
60 mg tablets: yellow, oval, biconvex, film-coated tablets, debossed with "L6" on one side and the Boehringer Ingelheim company symbol on the other side. Each tablet contains 60 mg of zongertinib.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1) ], HERNEXEOS can cause fetal harm when administered to a pregnant woman. There are no available data on the use of HERNEXEOS in pregnant women to inform a drug-associated risk. Oral administration of zongertinib to pregnant rats during the period of organogenesis caused structural abnormalities and alterations to growth at maternal exposures ≥ 19 times the human exposure based on AUC at the recommended dose [see Data ] . Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study, pregnant rats received oral doses of 10, 30, or 60 mg/kg/day of zongertinib during the period of organogenesis (gestation day 7 to 18). Zongertinib caused decreased fetal weights, delayed development of the urinary system, and kidney hydronephrosis at 60 mg/kg/day (approximately 19 times the human exposure based on AUC at the recommended dose). In an embryo-fetal development study in rabbits, there were no adverse embryo-fetal findings in pregnant animals administered oral doses of zongertinib up to 120 mg/kg/day (2.4 times the human exposure based on AUC at the recommended dose) during the period of organogenesis (gestation day 6 to 19).
A literature-based assessment of the effects on reproduction demonstrated that mice expressing catalytically inactive HER2 die at mid-gestation due to cardiac dysfunction.
Lactation
Risk Summary
There are no data on the presence of zongertinib or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with HERNEXEOS and for 2 weeks after the last dose.
Females and Males of Reproductive Potential
Based on findings from animal studies and its mechanism of action, HERNEXEOS can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1) ] .
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating HERNEXEOS.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with HERNEXEOS and for 2 weeks after the last dose.
Infertility
Females
Based on findings from animal studies, HERNEXEOS may impair fertility in females. The effects in female animals were reversible [see Nonclinical Toxicology 13.1 ] .
Males
Based on findings from animal studies, HERNEXEOS may impair fertility in males of reproductive potential. The effect on testes in animals was not reversible within a 4-week recovery period [see Nonclinical Toxicology 13.1 ] .
Pediatric Use
The safety and effectiveness of HERNEXEOS have not been established in pediatric patients.
Geriatric Use
Of the 292 patients with non-squamous NSCLC with HER2 (ERBB2) mutations who received HERNEXEOS in clinical studies, 47% were 65 years of age and older and 13% were 75 years and older. No overall differences in safety or effectiveness of HERNEXEOS were observed between older and younger adult patients.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Hepatotoxicity
HERNEXEOS can cause severe and life-threatening hepatotoxicity, including drug induced liver injury. In the pooled safety population [see Adverse Reactions (6.1) ], based on adverse reaction data, hepatotoxicity occurred in 27% of patients treated with HERNEXEOS. Grade 3 drug induced liver injury occurred in 1.4% and Grade 4 in 0.3% of patients treated with HERNEXEOS. Grade 3 hepatic failure occurred in 0.3% of patients treated with HERNEXEOS.
Based on laboratory data, 37% of patients treated with HERNEXEOS experienced increased alanine aminotransferase (ALT), including 4.9% Grade 3 and 1% Grade 4. Increased aspartate aminotransferase (AST) occurred in 31% of patients treated with HERNEXEOS, including 3.8% Grade 3 and 0.7% Grade 4. Increased bilirubin occurred in 20% of patients treated with HERNEXEOS, including 1% Grade 3 and 0.3% Grade 4.
HERNEXEOS was interrupted for an adverse reaction of hepatotoxicity in 8% of patients, the dose was reduced in 3.8% and permanently discontinued in 1%.
Monitor liver function tests including ALT, AST, and total bilirubin at baseline prior to administration of HERNEXEOS, every 2 weeks during the first 12 weeks, and then monthly thereafter as clinically indicated, with more frequent testing in patients who develop transaminase elevations. Interrupt, reduce the dose, or permanently discontinue HERNEXEOS based on the severity of the adverse reaction [see Dosage and Administration (2.3) ].
Left Ventricular Dysfunction
HERNEXEOS can cause severe left ventricular dysfunction. Left ventricular ejection fractions (LVEF) decrease occurred with anti-HER2 therapies, including HERNEXEOS. Treatment with HERNEXEOS has not been studied in patients with a history of clinically significant cardiac disease or LVEF less than 50% prior to initiation of treatment.
In the pooled safety population [see Adverse Reactions (6.1) ], LVEF decrease occurred in 6% of patients treated with HERNEXEOS, including 1.7% Grade 3. Two patients with Grade 3 LVEF decrease required permanent discontinuation of HERNEXEOS. The median time to onset of decreased LVEF was 12 weeks (range: 2.9 to 63 weeks).
Before initiating HERNEXEOS, evaluate LVEF and monitor at regular intervals during treatment and as clinically indicated. Interrupt, reduce the dose, or permanently discontinue HERNEXEOS based on the severity of the adverse reaction [see Dosage and Administration (2.3) ].
Interstitial Lung Disease/Pneumonitis
HERNEXEOS can cause severe and life-threatening interstitial lung disease (ILD)/pneumonitis.
In the pooled safety population [see Adverse Reactions (6.1) ], ILD/pneumonitis occurred in 2.1% of patients treated with HERNEXEOS. The median time to first onset of ILD/pneumonitis was 13 weeks (range: 1.4 to 65 weeks). One patient was able to resume therapy after resolution of pneumonitis. Two patients required permanent discontinuation and one patient died with unresolved pneumonitis > 30 days after discontinuing HERNEXEOS.
Monitor patients for new or worsening symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, fever). Interrupt, reduce the dose or permanently discontinue HERNEXEOS based on severity of confirmed ILD/pneumonitis [see Dosage and Administration (2.3) ].
Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, HERNEXEOS can cause fetal harm when administered to a pregnant woman. In an animal reproduction study, oral administration of zongertinib to pregnant rats during the period of organogenesis caused structural abnormalities and alterations to growth at maternal exposures ≥ 19 times the human exposure based on AUC at the recommended dose.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with HERNEXEOS and for 2 weeks after the last dose [see Use in Specific Populations (8.1 , 8.3) ].
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1) ]
- Left Ventricular Dysfunction [see Warnings and Precautions (5.2) ]
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.3) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The pooled safety population described in WARNINGS AND PRECAUTIONS reflects exposure to HERNEXEOS in 292 patients with unresectable or metastatic non-squamous NSCLC with HER2 (ERBB2) mutations who received HERNEXEOS as a single agent at 120 mg orally once daily until disease progression or unacceptable toxicity in Beamion LUNG-1 [see Clinical Studies (14) ] . Among 292 patients who received HERNEXEOS, 59% of patients were exposed for 6 months or longer and 28% were exposed for greater than one year. In this pooled safety population, the most common (> 20%) adverse reactions were diarrhea (54%), rash (28%), hepatotoxicity (27%), fatigue (25%), nausea (23%), musculoskeletal pain (21%), and upper respiratory tract infection (20%). The most common (≥ 2%) Grade 3 or 4 laboratory abnormalities were decreased lymphocytes (10%), increased alanine aminotransferase (6%), increased aspartate aminotransferase (4.5%), increased gamma glutamyl transferase (2.8%), decreased potassium (2.4%), and decreased neutrophils (2.4%).
Beamion LUNG-1
The safety of HERNEXEOS was evaluated in Beamion LUNG-1 in 177 patients with unresectable or metastatic non-squamous NSCLC with HER2 tyrosine kinase domain (TKD) mutations; 72 patients had not received prior treatment; 105 patients had received prior platinum-based chemotherapy, and out of those patients, 34 patients had received prior treatment with a HER2-directed antibody drug conjugate (ADC) [see Clinical Studies (14) ] . Patients received HERNEXEOS as a single agent at 120 mg once daily until disease progression or unacceptable toxicity. Among patients who received HERNEXEOS, 74% were exposed for 6 months or longer and 42% were exposed for greater than one year. The median age of patients who received HERNEXEOS was 63 years (range 30 to 88), 61% were female, 41% White, 48% Asian, and 0.6% Black or African American; 11% had unknown race data; 3.4% were of Hispanic or Latino ethnicity; and 38% had an Eastern Cooperative Oncology Group (ECOG) performance score of 0 and 62% had an ECOG performance score of 1.
Serious adverse reactions occurred in 36% of patients receiving HERNEXEOS. Serious adverse reactions in ≥ 2% of patients included pulmonary embolism (4%), dyspnea (3.4%), pneumonia (2.8%), hepatotoxicity, pleural effusion, and pericardial effusion (2.3%). Fatal adverse reactions occurred in one patient (0.6%) who received HERNEXEOS, due to pneumonia.
Permanent discontinuation of HERNEXEOS due to an adverse reaction occurred in 6% of patients. Adverse reactions which resulted in permanent discontinuation of HERNEXEOS were hepatotoxicity, decreased ejection fraction, anemia, increased blood alkaline phosphatase, diarrhea, dyspnea, increased gamma-glutamyl transferase, hemoptysis, pericardial effusion, pneumonitis, and pyrexia.
Dosage interruption of HERNEXEOS due to an adverse reaction occurred in 34% of patients. Adverse reactions which required dosage interruption in ≥ 2% of patients were hepatotoxicity, decreased ejection fraction, diarrhea, COVID-19, rash, and vomiting.
Dose reductions of HERNEXEOS due to adverse reactions occurred in 9% of patients. Adverse reactions which required dose reductions in ≥ 1% of patients were hepatotoxicity, decreased ejection fraction, and diarrhea.
Tables 3 and 4 summarize adverse reactions and laboratory abnormalities observed in Beamion LUNG-1.
| Adverse Reaction | HERNEXEOS N = 177 | |
|---|---|---|
| All Grades 1 % | Grade 3 or 4 % | |
| Events were graded using NCI CTCAE version 5.0. | ||
| 1 No Grade 4 or Grade 5 adverse reactions occurred. | ||
| •Grouped term. | ||
| Gastrointestinal Disorders | ||
| Diarrhea• | 56 | 2.8 |
| Nausea | 25 | 0.6 |
| Stomatitis• | 21 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||
| Rash• | 32 | 0.6 |
| Nail disorders• | 21 | 0.6 |
| Pruritus | 15 | 0 |
| General Disorders | ||
| Fatigue• | 26 | 0.6 |
| Headache | 16 | 0.6 |
| Respiratory, Thoracic and Mediastinal Disorders | ||
| Cough• | 23 | 0 |
| Musculoskeletal and Connective Tissue Disorders | ||
| Musculoskeletal pain• | 23 | 2.3 |
| Infections and Infestations | ||
| Upper respiratory tract infections• | 23 | 0 |
Clinically relevant adverse reactions in < 15% of patients who received HERNEXEOS included vomiting, dyspnea, dysgeusia, and dry skin.
| Laboratory Parameter | HERNEXEOS N = 177 | |
|---|---|---|
| All Grades 1 % | Grade 3 or 4 % | |
| Events were graded using NCI CTCAE version 5.0. | ||
| 1 No Grade 5 adverse reactions occurred. | ||
| Hematology | ||
| Lymphocytes decreased | 56 | 12 |
| Hemoglobin decreased | 37 | 1.1 |
| Leukocytes decreased | 35 | 1.1 |
| Activated partial thromboplastin time increased | 23 | 0 |
| Platelets decreased | 20 | 1.1 |
| Chemistry | ||
| Alanine aminotransferase increased | 41 | 7 |
| Aspartate aminotransferase increased | 35 | 5 |
| Creatinine kinase increased | 28 | 1.7 |
| Calcium decreased | 28 | 0.6 |
| Albumin decreased | 26 | 0 |
| Bilirubin increased | 25 | 1.1 |
| Lipase increased | 25 | 0 |
| Triglycerides increased | 25 | 0 |
| Sodium decreased | 22 | 0.6 |
| Bicarbonate decreased | 22 | 0 |
| Magnesium decreased | 21 | 1.1 |
| Potassium decreased | 21 | 1.7 |
| Alkaline phosphate increased | 20 | 1.7 |
DRUG INTERACTIONS
- Strong CYP3A Inducers : Avoid concomitant use with strong CYP3A inducers. If concomitant use cannot be avoided, increase HERNEXEOS dose. (7.1 )
- BCRP Substrates : Avoid concomitant use with certain BCRP substrates where minimal concentration increase may lead to serious adverse reactions and consider alternative therapies. If concomitant use cannot be avoided, monitor patients closely for adverse reactions and follow recommendations provided in the BCRP substrate approved product labeling. For other BCRP substrates, monitor for increased adverse reactions and adjust the dosages of those substrates as clinically appropriate. (7.2 )
Effects of Other Drugs on HERNEXEOS
Avoid concomitant use of HERNEXEOS with strong CYP3A inducers. If concomitant use cannot be avoided, increase HERNEXEOS dose as recommended [see Dosage and Administration (2.4) ].
Zongertinib is a CYP3A substrate. Strong CYP3A4 inducers decrease zongertinib exposure [see Clinical Pharmacology (12.3) ] , which may reduce effectiveness of HERNEXEOS.
Effects of HERNEXEOS on Other Drugs
BCRP Substrates
Avoid concomitant use of HERNEXEOS with certain BCRP substrates where minimal concentration changes may lead to serious adverse reactions. If coadministration cannot be avoided, monitor for increased adverse reactions and follow recommendations provided in the approved product labeling for the BCRP substrate. For other BCRP substrates, monitor for increased adverse reactions and adjust the dosages of those substrates as clinically appropriate.
Zongertinib is a BCRP inhibitor. HERNEXEOS increases exposure of BCRP substrates [see Clinical Pharmacology (12.3) ] , which may increase the risk of adverse reactions related to these substrates.
DESCRIPTION
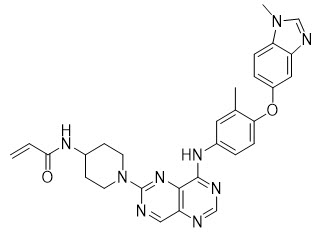
HERNEXEOS tablets for oral administration contain zongertinib, a kinase inhibitor. The chemical name of zongertinib is 2-Propenamide, N -[1-[8-[[3-methyl-4-[(1-methyl-1 H -benzimidazol-5-yl)oxy]phenyl] amino]pyrimido[5,4- d ]pyrimidin-2-yl]-4-piperidinyl]-. Its molecular formula is C 29 H 29 N 9 O 2 and the molecular weight is 535.6. The structural formula is:
Zongertinib is a yellow to dark yellow or orange solid. Zongertinib is slightly soluble at pH 1.2, and practically insoluble at pHs 3.6, 4.5, 5.4 and 6.8.
Each film-coated tablet of HERNEXEOS contains 60 mg of zongertinib and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose acetate succinate, mannitol, microcrystalline cellulose, and sodium stearyl fumarate. In addition, the film-coating contains the following inactive ingredients: ferric oxide (yellow), glycerol mono and dicaprylocaprate, polyvinyl alcohol, sodium lauryl sulfate, talc, and titanium dioxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Zongertinib is a kinase inhibitor of human epidermal growth factor receptor 2 (HER2). In vitro, zongertinib inhibited phosphorylation of HER2, downstream signaling of HER2 (phosphorylation of ERK), and proliferation of lung cancer cells harboring HER2 tyrosine kinase domain activating mutations. In vivo, zongertinib demonstrated anti-tumor activity in mouse xenograft models of NSCLC harboring HER2 tyrosine kinase domain activating mutations.
Pharmacodynamics
The exposure-response relationship and time-course of pharmacodynamic response of zongertinib have not been fully characterized.
Cardiac Electrophysiology
At 2.6 times the mean maximal concentration provided by the recommended dose of 120 mg, a mean increase in the QTc interval > 20 ms was not observed.
Pharmacokinetics
Zongertinib pharmacokinetics were observed at steady state in patients with advanced or metastatic solid tumors with HER2 aberrations at the approved recommended dosage and are presented as geometric mean (CV%), unless otherwise specified.
Zongertinib maximum concentration (C max,ss ) is 3.0 (37%) µmol/L and the total systemic exposure (AUC) is 34 (34%) µmol•h/L following HERNEXEOS 120 mg orally daily. Zongertinib C max and AUC increase in an approximately dose proportional manner across the dose range of 60 mg (0.5 times the approved recommended dosage) to 360 mg (3 times the approved recommended dosage). Zongertinib accumulation is approximately 1.5-fold for AUC and 1.3-fold for C max at the approved recommended dosage. Steady state is achieved within 2.5 days.
Absorption
Zongertinib median (min, max) time to maximum plasma concentration (T max ) is approximately 2 hours (min, max: 2, 6 hours). Zongertinib absolute oral bioavailability is 76%.
Effect of Food
No clinically significant differences in zongertinib C max and AUC were observed following administration of a single 240 mg dose (2 times the approved recommended dose) with a high-fat meal (approximately 1,000 calories, approximately 50% fat).
Distribution
Zongertinib plasma protein binding is > 99%. The apparent (oral) volume of distribution is 118 L (29%).
Elimination
Zongertinib effective half-life is 12 hours (21%) with an apparent (oral) clearance of 115 mL/min (31%).
Metabolism
Based on in vitro metabolite profiling, CYP-mediated oxidation pathways represent 48% to 62% (mainly CYP3A4 and CYP3A5), glucuronidation 13% to 25% (mainly UGT1A4), and glutathione conjugation 13% to 26% of total hepatic metabolism. Unchanged zongertinib represented the majority (75%) of total radioactivity in plasma.
Excretion
After a single oral dose of radiolabeled zongertinib 60 mg to healthy participants, approximately 93% of the dose was recovered in feces (31% unchanged) and 1.3% in urine (0.2% unchanged).
Specific Populations
The apparent volume of distribution and clearance of zongertinib increase with increasing body weight (34 to 122 kg).
No clinically significant differences in the pharmacokinetics of zongertinib were observed based on age (30 to 88 years), sex, race (36% White, 49% Asian, 1.3% Black/African American), mild renal impairment (eGFR 60 to < 90 mL/min) or mild hepatic impairment (AST > ULN and total bilirubin ≤ ULN; or total bilirubin > 1 to 1.5× ULN and any AST). The effect of moderate renal impairment (eGFR 30 to < 60 mL/min), severe renal impairment (eGFR 15 to < 30 mL/min), end-stage renal disease (eGFR < 15 mL/min), moderate hepatic impairment (total bilirubin > 1.5 to 3× ULN and any AST) or severe hepatic impairment (total bilirubin > 3× ULN and any AST) on the pharmacokinetics of zongertinib have not been studied.
Drug Interaction Studies
CYP3A Inducers: Zongertinib AUC decreased by 63% and C max decreased by 43% following concomitant use of carbamazepine (strong CYP3A inducer) 600 mg once daily for 7 days. The effect of concomitant use of moderate CYP3A inducers on zongertinib C max and AUC are unknown.
BCRP Substrates : Rosuvastatin (BCRP substrate) C max increased by 3-fold and AUC by 2.3-fold following concomitant use of a single dose of zongertinib 120 mg daily for 12 days.
Other Drugs: No clinically significant differences in zongertinib pharmacokinetics were observed when used concomitantly with strong CYP3A, P-gp, BCRP inhibitors and rabeprazole (proton pump inhibitor). No clinically significant differences in the pharmacokinetics of the following were observed when used concomitantly with zongertinib: midazolam (a CYP3A substrate), dabigatran (a P-gp substrate), metformin (a OCT2 and MATE1/2-K substrate), or repaglinide (a sensitive CYP2C8 substrate).
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies have not been conducted with zongertinib.
Mutagenesis
Zongertinib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay. Zongertinib was not genotoxic in an in vivo rat bone marrow micronucleus test and did not induce DNA breaks in a comet assay in liver or duodenum.
Impairment of Fertility
Dedicated animal fertility studies have not been conducted with zongertinib. In a 13-week repeat-dose toxicity study in rats, oral administration of zongertinib induced dose-dependent vacuolation in the testis at doses ≥ 10 mg/kg/day (≥ 5.7 times the human exposure based on AUC at the recommended dose), atrophy in the prostate gland at doses ≥ 30 mg/kg/day (≥ 8.4 times the human exposure based on AUC at the recommended dose), and atrophy in the uterus and hyperplasia/hyperkeratosis of the cervix and vagina at a dose of 90 mg/kg/day (approximately 17 times the human exposure based on AUC at the recommended dose). Findings in reproductive organs in female rats and prostate gland atrophy in male rats were reversible following a 4-week recovery period. Vacuolation in the testis of male rats was not reversible within a 4-week recovery period.
Animal Toxicology and/or Pharmacology
In a 4-week repeat-dose toxicology study in dogs, oral administration of zongertinib induced lesions of the oral mucosa (hard palate, buccal mucosa, tongue, lips) at doses ≥ 10 mg/kg/day (≥ 0.3 times the human exposure based on AUC at the recommended dose), which correlated with histologic erosion/ulcer and impaired food consumption. These findings were not present following a 4-week recovery period.
CLINICAL STUDIES
HERNEXEOS was evaluated in Beamion LUNG-1 (NCT04886804), a single arm, open-label, multi-center, multi-cohort trial. Eligible patients were required to have unresectable or metastatic NSCLC with HER2 (ERBB2) mutations. Patients with stable brain metastases were eligible to enroll. The study excluded patients who had a history of non-infectious interstitial lung disease/pneumonitis.
Patients received HERNEXEOS 120 mg orally once daily until disease progression or unacceptable toxicity. The major efficacy outcome measures were objective response rate (ORR) and duration of response (DOR) by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 as assessed by blinded independent central review (BICR).
Previously Untreated Unresectable or Metastatic Non-Squamous NSCLC
The efficacy population included 72 patients with unresectable or metastatic, non-squamous NSCLC with HER2 (ERBB2) tyrosine kinase domain (TKD) mutations based on prospective local testing, who had not received prior systemic therapy for advanced disease. Of those, tumor tissue samples from 60% (43/72) of patients were retrospectively tested using Oncomine™ Dx Target Test (Life Technologies Corporation, Tissue-test). While 86% (37/43) of samples were positive for HER2 (ERBB2) TKD mutations; 14% (6/43) were unevaluable.
The baseline demographic and disease characteristics of the efficacy population were: 67 years (range: 35 to 88); 50% female, 47% Asian, 42% White, 1.4% Black or African American; 10% had unknown race data; 6% were of Hispanic or Latino ethnicity; 44% Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0 and 56% ECOG PS 1; 65% never smoked; 100% had metastatic disease; and 31% had brain metastases.
Efficacy results are summarized in Table 5.
| Efficacy Parameter | HERNEXEOS N = 72 |
|---|---|
| Abbreviation: CI = Confidence Interval, +: Ongoing response. | |
| 1 Based on Wilson confidence interval. | |
| 2 Based on observed duration of response. | |
| Objective Response Rate (ORR), % (95% CI) 1 | 76 (65, 85) |
| Complete response, % | 11 |
| Partial response, % | 65 |
| Duration of Response (DOR) | N = 55 |
| Range, months | 1.4, 18+ |
| DOR ≥ 6 months, 2 % | 64 |
| DOR ≥ 12 months, 2 % | 44 |
Previously Treated Unresectable or Metastatic Non-Squamous NSCLC
The efficacy population included 71 patients with unresectable or metastatic, non-squamous NSCLC with HER2 (ERBB2) TKD mutations based on prospective local testing. Of those, tumor tissue samples from 52% (37/71) of patients were retrospectively tested using Oncomine™ Dx Target Test (Life Technologies Corporation, Tissue-test). While 84% (31/37) of samples were positive for HER2 (ERBB2) TKD mutations, 2.7% (1/37) did not have HER2 (ERBB2) TKD mutations identified, and 13.5% (5/37) were unevaluable.
The baseline demographic and disease characteristics of the efficacy population were: 62 years (range: 30 to 80); 70% female, 55% Asian, 35% White, 0% Black or African American; 10% had unknown race data; 1.4% were of Hispanic or Latino ethnicity; 39% Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0 and 61% ECOG PS 1; 65% never smoked; 100% metastatic disease; and 37% with brain metastases. The median number of prior therapies was 1 (range: 1 to 10); 100% of patients had prior platinum therapy and 78% had prior treatment with anti-PD-1/PD-L1 antibody. No patient had received previous treatment with a HER2-targeted tyrosine kinase inhibitor (TKI) or HER2-targeted antibody-drug conjugate (ADC).
Efficacy results are summarized in Table 6.
| Efficacy Parameter | HERNEXEOS N = 71 |
|---|---|
| Abbreviation: CI = Confidence Interval, +: Ongoing response. | |
| 1 Based on Wilson confidence interval. | |
| 2 Based on observed duration of response. | |
| Objective Response Rate (ORR), % (95% CI) 1 | 75 (63, 83) |
| Complete response, % | 6 |
| Partial response, % | 69 |
| Duration of Response (DOR) | N = 53 |
| Range, months | 1.3+, 15+ |
| DOR ≥ 6 months, 2 % | 58 |
Among the 71 patients, 5 patients had measurable CNS metastases at baseline as assessed by BICR and had not received radiation therapy to the brain within 2 months prior to treatment with HERNEXEOS. Based on Response Assessment in Neuro-Oncology Brain Metastases (RANO-BM) criteria per BICR, responses were observed in 3 patients.
HERNEXEOS was also evaluated in 34 patients with unresectable or metastatic HER2 (ERBB2) TKD mutation-positive non-squamous NSCLC who had received previous treatment with platinum-based chemotherapy and a HER2-targeted ADC. Eligibility criteria were otherwise similar to the efficacy population described above. The median age was 58 years (range: 31 to 85); 65% female, 35% Asian, 50% White, 0% Black or African American; 15% had unknown race data; and 2.9% were of Hispanic or Latino ethnicity. Baseline ECOG performance status was 0 (21%) or 1 (79%); 65% never smoked; 100% of patients had metastatic disease and 74% had brain metastases. The median number of prior therapies was 3 (range: 1 to 8); 100% of patients had prior platinum therapy and 77% had prior treatment with anti-PD-1/PD-L1 antibody; 2.9% of patients had received previous treatment with a HER2-targeted TKI. Confirmed ORR by RECIST v1.1 based on BICR was 44% (95% CI 29, 61), with 2.9% of patients having a complete response. Median DOR was 5.4 months (95% CI 2.8, not estimable), and 27% of responders had an observed DOR ≥ 6 months.
HOW SUPPLIED/STORAGE AND HANDLING
60 mg tablets: yellow, oval, biconvex, film-coated, debossed with "L6" on one side and the Boehringer Ingelheim company symbol on the other side. They are packaged in a bottle containing two silica gel desiccants and with a child-resistant closure, available as follows:
| Carton containing one bottle of 30 tablets each | NDC: 0597-9257-59 |
| Carton containing one bottle of 60 tablets each | NDC: 0597-9257-86 |
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F), excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Store in the original container to protect from moisture. Keep the bottle tightly closed. Do not remove the desiccants. Once opened, use within 3 months. Discard any unused tablets 3 months after opening the bottle.
Mechanism of Action
Zongertinib is a kinase inhibitor of human epidermal growth factor receptor 2 (HER2). In vitro, zongertinib inhibited phosphorylation of HER2, downstream signaling of HER2 (phosphorylation of ERK), and proliferation of lung cancer cells harboring HER2 tyrosine kinase domain activating mutations. In vivo, zongertinib demonstrated anti-tumor activity in mouse xenograft models of NSCLC harboring HER2 tyrosine kinase domain activating mutations.