Get your patient on Arynta (Lisdexamfetamine Dimesylate Oral)
Arynta prescribing information
WARNING: ABUSE, MISUSE, AND ADDICTION
ARYNTA has a high potential for abuse and misuse, which can lead to the development of a substance use disorder, including addiction. Misuse and abuse of CNS stimulants, including ARYNTA, can result in overdose and death [see Overdosage (10 )] , and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
Before prescribing ARYNTA, assess each patient's risk for abuse, misuse, and addiction. Educate patients and their families about these risks, proper storage of the drug, and proper disposal of any unused drug. Throughout ARYNTA treatment, reassess each patient's risk of abuse, misuse, and addiction and frequently monitor for signs and symptoms of abuse, misuse, and addiction [see Warnings and Precautions (5.1 ), Drug Abuse and Dependence (9.2 )].
INDICATIONS AND USAGE
ARYNTA is indicated for the treatment of:
- Attention Deficit Hyperactivity Disorder (ADHD) in adults and pediatric patients 6 years and older [see Clinical Studies (14.1 )]
- Moderate to severe binge eating disorder (BED) in adults [see Clinical Studies (14.2 )] .
Limitations of Use:
- The use of ARYNTA is not recommended in pediatric patients younger than 6 years of age because they had higher plasma exposure and a higher incidence of adverse reactions (e.g., weight loss) than patients 6 years and older at the same dosage [see Warnings and Precautions (5.5 ), Use in Specific Populations (8.4 )] .
- ARYNTA is not indicated or recommended for weight loss. Use of other sympathomimetic drugs for weight loss has been associated with serious cardiovascular adverse events. The safety and effectiveness of ARYNTA for the treatment of obesity have not been established [see Warnings and Precautions (5.2 )] .
DOSAGE AND ADMINISTRATION
| Indicated Population | Initial Dose | Titration Schedule | Recommended Dose | Maximum Dose |
| ADHD (Adults and pediatric patients 6 years and older) (2.2 ) | 30 mg every morning | 10 mg or 20 mg weekly | 30 mg to 70 mg per day | 70 mg per day |
| BED (Adults) (2.3 ) | 30 mg every morning | 20 mg weekly | 50 mg to 70 mg per day | 70 mg per day |
• Prior to treatment, assess for presence of cardiac disease (2.4 ) • Severe renal impairment: Maximum dose is 50 mg/day (2.5 ) • End stage renal disease (ESRD): Maximum dose is 30 mg/day (2.5 )
Pretreatment Screening
Prior to treating patients with ARYNTA, assess: • for the presence of cardiac disease (i.e., perform a careful history, family history of sudden death or ventricular arrhythmia, and physical exam) [see Warnings and Precautions (5.2 )] . • the family history and clinically evaluate patients for motor or verbal tics or Tourette's syndrome before initiating ARYNTA [see Warnings and Precautions (5.8 )] .
General Administration Information
Take ARYNTA orally in the morning with or without food; avoid afternoon doses because of the potential for insomnia. Instruct caregivers/patients to read the ‘Instructions for Use’ for complete administration instructions. • Use the oral dosing syringe and bottle adapter provided with ARYNTA. • Ensure that the bottle adapter is firmly inserted into the bottle before first use and keep the adapter in place for the duration of the usage of the bottle. • Throw away (discard) any remaining ARYNTA 30 days after first opening the bottle.
Dosage for Treatment of ADHD
The recommended starting dosage in adults and pediatric patients 6 years and older is 30 mg once daily in the morning. Dosage may be adjusted in increments of 10 mg or 20 mg at approximately weekly intervals up to maximum recommended dosage of 70 mg once daily [see Clinical Studies (14.1 )] .
Dosage for Treatment of Moderate to Severe BED in Adults
The recommended starting dosage in adults is 30 mg once daily to be titrated in increments of 20 mg at approximately weekly intervals to achieve the recommended target dose of 50 mg to 70 mg once daily. The maximum recommended dosage is 70 mg once daily [see Clinical Studies (14.2 )]. Discontinue ARYNTA if binge eating does not improve.
Dosage in Patients with Renal Impairment
In patients with severe renal impairment (GFR 15 to < 30 mL/min/1.73 m 2 ), the maximum dosage should not exceed 50 mg once daily. In patients with end stage renal disease (ESRD, GFR < 15 mL/min/1.73 m 2 ), the maximum recommended dosage is 30 mg once daily [see Use in Specific Populations (8.6 )] .
Dosage Modifications due to Drug Interactions
Agents that alter urinary pH can impact urinary excretion and alter blood levels of amphetamine. Acidifying agents (e.g., ascorbic acid) decrease blood levels, while alkalinizing agents (e.g., sodium bicarbonate) increase blood levels. Adjust ARYNTA dosage accordingly [see Drug Interactions (7.1 )] .
DOSAGE FORMS AND STRENGTHS
ARYNTA oral solution 10 mg/mL is supplied as a clear colorless solution.
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ADHD medications during pregnancy. Healthcare providers are encouraged to register patients by calling the National Pregnancy Registry for Psychostimulants at 1-866-961-2388 or visiting online at https://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/adhd-medications/.
Risk Summary
The limited available data from published literature and postmarketing reports on use of ARYNTA in pregnant women are not sufficient to inform a drug-associated risk for major birth defects and miscarriage. Adverse pregnancy outcomes, including premature delivery and low birth weight, have been seen in infants born to mothers dependent on amphetamines [ see Clinical Considerations ]. In animal reproduction studies, lisdexamfetamine dimesylate (a prodrug of d-amphetamine) had no effects on embryo-fetal morphological development or survival when administered orally to pregnant rats and rabbits throughout the period of organogenesis. Pre- and postnatal studies were not conducted with lisdexamfetamine dimesylate. However, amphetamine (d- to l- ratio of 3:1) administration to pregnant rats during gestation and lactation caused a decrease in pup survival and a decrease in pup body weight that correlated with a delay in developmental landmarks at clinically relevant doses of amphetamine. In addition, adverse effects on reproductive performance were observed in pups whose mothers were treated with amphetamine. Long-term neurochemical and behavioral effects have also been reported in animal developmental studies using clinically relevant doses of amphetamine [see Data]. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. Clinical Considerations Fetal/Neonatal Adverse Reactions Amphetamines, such as ARYNTA, cause vasoconstriction and thereby may decrease placental perfusion. In addition, amphetamines can stimulate uterine contractions increasing the risk of premature delivery. Infants born to amphetamine-dependent mothers have an increased risk of premature delivery and low birth weight. Monitor infants born to mothers taking amphetamines for symptoms of withdrawal such as feeding difficulties, irritability, agitation, and excessive drowsiness. Data Animal Data Lisdexamfetamine dimesylate had no apparent effects on embryo-fetal morphological development or survival when administered orally to pregnant rats and rabbits throughout the period of organogenesis at doses of up to 40 and 120 mg/kg/day, respectively. These doses are approximately 5.5 and 33 times, respectively, the maximum recommended human dose (MRHD) of 70 mg/day given to adults, on a mg/m2 body surface area basis. A study was conducted with amphetamine (d- to l- enantiomer ratio of 3:1) in which pregnant rats received daily oral doses of 2, 6, and 10 mg/kg from gestation day 6 to lactation day 20. All doses caused hyperactivity and decreased weight gain in the dams. A decrease in pup survival was seen at all doses. A decrease in pup body weight was seen at 6 and 10 mg/kg which correlated with delays in developmental landmarks, such as preputial separation and vaginal opening. Increased pup locomotor activity was seen at 10 mg/kg on day 22 postpartum but not at 5 weeks postweaning. When pups were tested for reproductive performance at maturation, gestational weight gain, number of implantations, and number of delivered pups were decreased in the group whose mothers had been given 10 mg/kg. A number of studies from the literature in rodents indicate that prenatal or early postnatal exposure to amphetamine (d- or d, l-) at doses similar to those used clinically can result in long-term neurochemical and behavioral alterations. Reported behavioral effects include learning and memory deficits, altered locomotor activity, and changes in sexual function.
Lactation
Risk Summary Lisdexamfetamine is a pro-drug of dextroamphetamine. Based on limited case reports in published literature, amphetamine ( d -or d , l -) is present in human milk, at relative infant doses of 2% to 13.8% of the maternal weight-adjusted dosage and a milk/plasma ratio ranging between 1.9 and 7.5. There are no reports of adverse effects on the breastfed infant. Long-term neurodevelopmental effects on infants from amphetamine exposure are unknown. It is possible that large dosages of dextroamphetamine might interfere with milk production, especially in women whose lactation is not well established. Because of the potential for serious adverse reactions in nursing infants, including serious cardiovascular reactions, blood pressure and heart rate increase, suppression of growth, and peripheral vasculopathy, advise patients that breastfeeding is not recommended during treatment with ARYNTA.
Pediatric Use
The safety and effectiveness of ARYNTA have not been established in pediatric patients below the age of 6 years.
ADHD Safety and effectiveness of ARYNTA have been established in pediatric patients with ADHD ages 6 to 17 years [see Dosage and Administration (2.3 ), Adverse Reactions (6.1 ), Clinical Pharmacology (12.3 ), and Clinical Studies (14.1 )]. Safety and efficacy of lisdexamfetamine dimesylate were evaluated in a double-blind, randomized, parallel-group, placebo-controlled, fixed-dose study in pediatric patients ages 4 to 5 years with ADHD, followed by a 1-year open-label extension study. In these studies, patients experienced elevated rates of adverse reactions, including weight loss, decreased BMI, decreased appetite, insomnia, infections (upper respiratory and nasopharyngitis), irritability, and affect lability. With the same lisdexamfetamine dimesylate dose, mean steady state exposure of dextroamphetamine was approximately 44% higher in pediatric patients ages 4 to 5 years compared to the pediatric patients ages 6 to 11 years. BED Safety and effectiveness of ARYNTA have not been established in pediatric patients with BED less than 18 years of age. Growth Suppression Growth should be monitored during treatment with stimulants, including ARYNTA, and pediatric patients who are not growing or gaining weight as expected may need to have their treatment interrupted [see Warnings and Precautions (5.5 ) and Adverse Reactions (6.1 )]. Juvenile Animal Data Studies conducted in juvenile rats and dogs at clinically relevant doses showed growth suppression that partially or fully reversed in dogs and female rats but not in male rats after a four-week drug-free recovery period. A study was conducted in which juvenile rats received oral doses of 4, 10, or 40 mg/kg/day of lisdexamfetamine dimesylate from day 7 to day 63 of age. These doses are approximately 0.3, 0.7, and 3 times the maximum recommended human daily dose of 70 mg on a mg/m 2 basis for a child. Dose-related decreases in food consumption, bodyweight gain, and crown-rump length were seen; after a four-week drug-free recovery period, bodyweights and crown-rump lengths had significantly recovered in females but were still substantially reduced in males. Time to vaginal opening was delayed in females at the highest dose, but there were no drug effects on fertility when the animals were mated beginning on day 85 of age. In a study in which juvenile dogs received lisdexamfetamine dimesylate for 6 months beginning at 10 weeks of age, decreased bodyweight gain was seen at all doses tested (2, 5, and 12 mg/kg/day, which are approximately 0.5, 1, and 3 times the maximum recommended human daily dose on a mg/m 2 basis for a child). This effect partially or fully reversed during a four-week drug-free recovery period.
Geriatric Use
Clinical studies of lisdexamfetamine dimesylate did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience and pharmacokinetic data [see Clinical Pharmacology (12.3 )] have not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should start at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Renal Impairment
Due to reduced clearance in patients with severe renal impairment (GFR 15 to <30 mL/min/1.73 m 2 ), the maximum dose should not exceed 50 mg/day. The maximum recommended dose in ESRD (GFR <15 mL/min/1.73 m 2 ) patients is 30 mg/day [see Clinical Pharmacology (12.3 )]. Lisdexamfetamine and d-amphetamine are not dialyzable.
CONTRAINDICATIONS
ARYNTA is contraindicated in patients with: • Known hypersensitivity to amphetamine products or other ingredients of ARYNTA. Anaphylactic reactions, Stevens-Johnson Syndrome, angioedema, and urticaria have been observed in postmarketing reports [see Adverse Reactions (6.2 )]. • Patients taking monoamine oxidase inhibitors (MAOIs), or within 14 days of stopping MAOIs (including MAOIs such as linezolid or intravenous methylene blue), because of an increased risk of hypertensive crisis [see Warnings and Precautions (5.7 ) and Drug Interactions (7.1 )].
WARNINGS AND PRECAUTIONS
• Risks to Patients with Serious Cardiac Disease : Avoid use in patients with known structural cardiac abnormalities, cardiomyopathy, serious cardiac arrhythmia, coronary artery disease, or other serious cardiac disease (5.2 ) • Increased Blood Pressure and Heart Rate : Monitor blood pressure and pulse. (5.3 ) • Psychiatric Adverse Reactions : Prior to initiating ARYNTA, screen patients for risk factors for developing a manic episode. If new psychotic or manic symptoms occur, consider discontinuing ARYNTA. (5.4 ) • Long-Term Suppression of Growth in Pediatric Patients : Closely monitor growth (height and weight) in pediatric patients. Pediatric patients not growing or gaining height or weight as expected may need to have their treatment interrupted. (5.5 ) • Peripheral Vasculopathy, including Raynaud's phenomenon : Careful observation for digital changes is necessary during ARYNTA treatment. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for patients who develop signs or symptoms of peripheral vasculopathy. (5.6 ) • Serotonin Syndrome : Increased risk when co-administered with serotonergic agents (e.g., SSRIs, SNRIs, triptans), but also during overdosage situations. If it occurs, discontinue ARYNTA and initiate supportive treatment. (4 , 5.7 , 10 ) • Motor and Verbal Tics, and Worsening of Tourette's Syndrome : Before initiating ARYNTA, assess the family history and clinically evaluate patients for tics or Tourette's syndrome. Regularly monitor patients for the emergence or worsening of tics or Tourette's syndrome. Discontinue treatment if clinically appropriate. (5.8 )
Abuse, Misuse, and Addiction
ARYNTA has a high potential for abuse and misuse. The use of ARYNTA exposes individuals to the risks of abuse and misuse, which can lead to the development of a substance use disorder, including addiction. ARYNTA can be diverted for non-medical use into illicit channels or distribution [see Drug Abuse and Dependence (9.2 )] . Misuse and abuse of CNS stimulants, including ARYNTA, can result in overdose and death [see Overdosage (10 )] , and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection. Before prescribing ARYNTA, assess each patient's risk for abuse, misuse, and addiction. Educate patients and their families about these risks and proper disposal of any unused drug. Advise patients to store ARYNTA in a safe place, preferably locked, and instruct patients to not give ARYNTA to anyone else. Throughout ARYNTA treatment, reassess each patient's risk of abuse, misuse, and addiction and frequently monitor for signs and symptoms of abuse, misuse, and addiction.
Risks to Patients with Serious Cardiac Disease
Sudden death has been reported in patients with structural cardiac abnormalities or other serious cardiac disease who were treated with CNS stimulants at the recommended ADHD dosage. Avoid ARYNTA use in patients with known structural cardiac abnormalities, cardiomyopathy, serious cardiac arrhythmia, coronary artery disease, or other serious cardiac disease.
Increased Blood Pressure and Heart Rate
CNS stimulants cause an increase in blood pressure (mean increase about 2 to 4 mm Hg) and heart rate (mean increase about 3 to 6 bpm). Some patients may have larger increases. Monitor all ARYNTA -treated patients for potential tachycardia and hypertension.
Psychiatric Adverse Reactions
Exacerbation of Pre-existing Psychosis CNS stimulants may exacerbate symptoms of behavior disturbance and thought disorder in patients with a pre-existing psychotic disorder. Induction of a Manic Episode in Patients with Bipolar Disorder CNS stimulants may induce a manic or mixed episode. Prior to initiating ARYNTA treatment, screen patients for risk factors for developing a manic episode (e.g., comorbid or history of depressive symptoms or a family history of suicide, bipolar disorder, and depression). New Psychotic or Manic Symptoms CNS stimulants, at the recommended dosage, may cause psychotic or manic symptoms (e.g., hallucinations, delusional thinking, or mania) in patients without a prior history of psychotic illness or mania. In a pooled analysis of multiple short-term, placebo-controlled studies of CNS stimulants, psychotic or manic symptoms occurred in approximately 0.1% of CNS stimulant-treated patients compared to 0% of placebo-treated patients. If such symptoms occur, consider discontinuing ARYNTA.
Long-Term Suppression of Growth in Pediatric Patients
ARY NTA is not approved for use and is not recommended in pediatric patients below 6 years of age [see Use in Specific Populations (8.4 )] .
CNS stimulants have been associated with weight loss and slowing of growth rate in pediatric patients. In a 4-week, placebo-controlled trial of ARYNTA in pediatric patients ages 6 to 12 years old with ADHD, there was a dose-related decrease in weight in the ARYNTA groups compared to weight gain in the placebo group. Additionally, in studies of another stimulant, there was slowing of the increase in height [see Adverse Reactions (6.1 )] . Closely monitor growth (weight and height) in ARYNTA-treated pediatric patients. Patients who are not growing or gaining height or weight as expected may need to have their treatment interrupted.
Peripheral Vasculopathy, including Raynaud's Phenomenon
CNS stimulants, including ARYNTA, used to treat ADHD are associated with peripheral vasculopathy, including Raynaud's phenomenon. Signs and symptoms are usually intermittent and mild; however, sequelae have included digital ulceration and/or soft tissue breakdown. Effects of peripheral vasculopathy, including Raynaud's phenomenon, were observed in post-marketing reports and at the therapeutic dosages of CNS stimulants in all age groups throughout the course of treatment. Signs and symptoms generally improved after dosage reduction or discontinuation of the CNS stimulant. Careful observation for digital changes is necessary during ARYNTA treatment. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for ARYNTA-treated patients who develop signs or symptoms of peripheral vasculopathy.
Serotonin Syndrome
Serotonin syndrome, a potentially life-threatening reaction, may occur when amphetamines are used in combination with other drugs that affect the serotonergic neurotransmitter systems such as monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), serotonin norepinephrine reuptake inhibitors (SNRIs), triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, and St. John's Wort [see Drug Interactions (7.1 )] . The co-administration with cytochrome P450 2D6 (CYP2D6) inhibitors may also increase the risk with increased exposure to the active metabolite of ARYNTA (dextroamphetamine). In these situations, consider an alternative non-serotonergic drug or an alternative drug that does not inhibit CYP2D6 [see Drug Interactions (7.1 )] . Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Concomitant use of ARYNTA with MAOI drugs is contraindicated [see Contraindications (4 )] . Discontinue treatment with ARYNTA and any concomitant serotonergic agents immediately if symptoms of serotonin syndrome occur, and initiate supportive symptomatic treatment. If concomitant use of ARYNTA with other serotonergic drugs or CYP2D6 inhibitors is clinically warranted, initiate ARYNTA with lower doses, monitor patients for the emergence of serotonin syndrome during drug initiation or titration, and inform patients of the increased risk for serotonin syndrome.
Motor and Verbal Tics, and Worsening of Tourette's Syndrome
CNS stimulants, including amphetamine, have been associated with the onset or exacerbation of motor and verbal tics. Worsening of Tourette's syndrome has also been reported [see Adverse Reactions (6.2 )] . Before initiating ARYNTA, assess the family history and clinically evaluate patients for tics or Tourette's syndrome. Regularly monitor ARYNTA -treated patients for the emergence or worsening of tics or Tourette's syndrome, and discontinue treatment if clinically appropriate.
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling: • Known hypersensitivity to amphetamine products or other ingredients of ARYNTA [see Contraindications (4 )] • Hypertensive Crisis When Used Concomitantly with Monoamine Oxidase Inhibitors [see Contraindications (4 ) and Drug Interactions (7.1 )] • Abuse, Misuse, and Addiction [see Boxed Warning, Warnings and Precautions (5.1 ), and Drug Abuse and Dependence (9.2 , 9.3 )] • Risks to Patients with Serious Cardiac Disease [see Warnings and Precautions (5.2 )] • Increased Blood Pressure and Heart Rate [see Warnings and Precautions (5.3 )] • Psychiatric Adverse Reactions [see Warnings and Precautions (5.4 )] • Long-Term Suppression of Growth in Pediatric Patients [see Warnings and Precautions (5.5 )] • Peripheral Vasculopathy, including Raynaud's phenomenon [see Warnings and Precautions (5.6 )] • Serotonin Syndrome [see Warnings and Precautions (5.7 )] • Motor and Verbal Tics, and Worsening of Tourette's Syndrome [see Warnings and Precautions (5.8 )]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. Attention Deficit Hyperactivity Disorder The safety data in this section is based on data from the 4-week controlled parallel-group clinical studies of Lisdexamfetamine Dimesylate in pediatric and adult patients with ADHD [see Clinical Studies (14.1 )]. Adverse Reactions Associated with Discontinuation of Treatment in ADHD Clinical Trials In the controlled trial in pediatric patients ages 6 to 12 years (Study 1), 8% (18/218) of ARYNTA-treated patients discontinued due to adverse reactions compared to 0% (0/72) of placebo-treated patients. The most frequently reported adverse reactions (1% or more and twice rate of placebo) were ECG voltage criteria for ventricular hypertrophy, tic, vomiting, psychomotor hyperactivity, insomnia, decreased appetite and rash [2 instances for each adverse reaction, i.e., 2/218 (1%)]. Less frequently reported adverse reactions (less than 1% or less than twice rate of placebo) included abdominal pain upper, dry mouth, weight decreased, dizziness, somnolence, logorrhea, chest pain, anger and hypertension. In the controlled trial in pediatric patients ages 13 to 17 years (Study 4), 3% (7/233) of ARYNTA-treated patients discontinued due to adverse reactions compared to 1% (1/77) of placebo-treated patients. The most frequently reported adverse reactions (1% or more and twice rate of placebo) were decreased appetite (2/233; 1%) and insomnia (2/233; 1%). Less frequently reported adverse reactions (less than 1% or less than twice rate of placebo) included irritability, dermatillomania, mood swings, and dyspnea. In the controlled adult trial (Study 7), 6% (21/358) of ARYNTA-treated patients discontinued due to adverse reactions compared to 2% (1/62) of placebo-treated patients. The most frequently reported adverse reactions (1% or more and twice rate of placebo) were insomnia (8/358; 2%), tachycardia (3/358; 1%), irritability (2/358; 1%), hypertension (4/358; 1%), headache (2/358; 1%), anxiety (2/358; 1%), and dyspnea (3/358; 1%). Less frequently reported adverse reactions (less than 1% or less than twice rate of placebo) included palpitations, diarrhea, nausea, decreased appetite, dizziness, agitation, depression, paranoia and restlessness. Adverse Reactions Occurring at an Incidence of ≥5% or More Among ARYNTA Treated Patients with ADHD in Clinical Trials The most common adverse reactions (incidence ≥5% and at a rate at least twice placebo) reported in pediatric patients ages 6 to 17 years, and/or adults were anorexia, anxiety, decreased appetite, decreased weight, diarrhea, dizziness, dry mouth, irritability, insomnia, nausea, upper abdominal pain, and vomiting. Adverse Reactions Occurring at an Incidence of 2% or More Among ARYNTA Treated Patients with ADHD in Clinical Trials Adverse reactions reported in the controlled trials in pediatric patients ages, 6 to 12 years (Study 1), pediatric patients ages 13 to 17 years (Study 4), and adult patients (Study 7) treated with ARYNTA or placebo are presented in Tables 1, 2 and 3 below.
Table 1 Adverse Reactions Reported by 2% or More of Pediatric Patients Ages 6 to 12 Years with ADHD Taking Lisdexamfetamine Dimesylate and Greater than or Equal to Twice the Incidence in Patients Taking Placebo in a 4-Week Clinical Trial (Study 1)
| Lisdexamfetamine Dimesylate (n=218) | Placebo (n=72) | |
| Decreased Appetite | 39% | 4% |
| Insomnia | 22% | 3% |
| Abdominal Pain Upper | 12% | 6% |
| Irritability | 10% | 0% |
| Vomiting | 9% | 4% |
| Weight Decreased | 9% | 1% |
| Nausea | 6% | 3% |
| Dry Mouth | 5% | 0% |
| Dizziness | 5% | 0% |
| Affect lability | 3% | 0% |
| Rash | 3% | 0% |
| Pyrexia | 2% | 1% |
| Somnolence | 2% | 1% |
| Tic | 2% | 0% |
| Anorexia | 2% | 0% |
Table 2 Adverse Reactions Reported by 2% or More of Pediatric Patients Ages 13 to 17 Years with ADHD Taking Lisdexamfetamine Dimesylate and Greater than or Equal to Twice the Incidence in Patients Taking Placebo in a 4-Week Clinical Trial (Study 4)
| L isdexamfetamine Dimesylate (n=233) | Placebo (n=77) | |
| Decreased Appetite | 34% | 3% |
| Insomnia | 13% | 4% |
| Weight Decreased | 9% | 0% |
| Dry Mouth | 4% | 1% |
| Palpitations | 2% | 1% |
| Anorexia | 2% | 0% |
| Tremor | 2% | 0% |
Table 3. Adverse Reactions Reported by 2% or More of Adult Patients with ADHD Taking Lisdexamfetamine Dimesylate and Greater than or Equal to Twice the Incidence in Patients Taking Placebo in a 4-Week Clinical Trial (Study 7)
| Lisdexamfetamine Dimesylate (n=358) | Placebo (n=62) | |
| Decreased Appetite | 27% | 2% |
| Insomnia | 27% | 8% |
| Dry Mouth | 26% | 3% |
| Diarrhea | 7% | 0% |
| Nausea | 7% | 0% |
| Anxiety | 6% | 0% |
| Anorexia | 5% | 0% |
| Feeling Jittery | 4% | 0% |
| Agitation | 3% | 0% |
| Increased Blood Pressure | 3% | 0% |
| Hyperhidrosis | 3% | 0% |
| Restlessness | 3% | 0% |
| Decreased Weight | 3% | 0% |
| Dyspnea | 2% | 0% |
| Increased Heart Rate | 2% | 0% |
| Tremor | 2% | 0% |
| Palpitations | 2% | 0% |
In addition, in the adult population erectile dysfunction was observed in 2.6% of males on lisdexamfetamine dimesylate and 0% on placebo; decreased libido was observed in 1.4% of subjects on lisdexamfetamine dimesylate and 0% on placebo.
Weight Loss and Slowing Growth Rate in Pediatric Patients with ADHD In a controlled trial of lisdexamfetamine dimesylate in pediatric patients ages 6 to 12 years (Study 1), mean weight loss from baseline after 4 weeks of therapy was -0.9, -1.9, and -2.5 pounds, respectively, for patients receiving 30 mg, 50 mg, and 70 mg of lisdexamfetamine dimesylate, compared to a 1 pound weight gain for patients receiving placebo. Higher doses were associated with greater weight loss with 4 weeks of treatment. Careful follow-up for weight in pediatric patients ages 6 to 12 years who received lisdexamfetamine dimesylate over 12 months suggests that consistently medicated pediatric patients (i.e., treatment for 7 days per week throughout the year) have a slowing in growth rate, measured by body weight as demonstrated by an age- and sex-normalized mean change from baseline in percentile, of -13.4 over 1 year (average percentiles at baseline and 12 months were 60.9 and 47.2, respectively). In a 4-week controlled trial of lisdexamfetamine dimesylate in pediatric patients ages 13 to 17 years, mean weight loss from baseline to endpoint was -2.7, -4.3, and -4.8 lbs., respectively, for patients receiving 30 mg, 50 mg, and 70 mg of lisdexamfetamine dimesylate, compared to a 2.0 pound weight gain for patients receiving placebo. Careful follow-up of weight and height in pediatric patients ages 7 to 10 years who were randomized to either methylphenidate or non-medication treatment groups over 14 months, as well as in naturalistic subgroups of newly methylphenidate-treated and non-medication treated pediatric patients over 36 months (to the ages of 10 to 13 years), suggests that consistently medicated pediatric patients ages 7 to 13 years (i.e., treatment for 7 days per week throughout the year) have a temporary slowing in growth rate (on average, a total of about 2 cm less growth in height and 2.7 kg less growth in weight over 3 years), without evidence of growth rebound during this period of development. In a controlled trial of amphetamine (d- to l-enantiomer ratio of 3:1) in pediatric patients ages 13 to 17 years, mean weight change from baseline within the initial 4 weeks of therapy was -1.1 pounds and -2.8 pounds, respectively, for patients receiving 10 mg and 20 mg of amphetamine. Higher doses were associated with greater weight loss within the initial 4 weeks of treatment [see Warnings and Precautions (5.5 )]. Weight Loss in Adults with ADHD In the controlled adult trial (Study 7), mean weight loss after 4 weeks of therapy was 2.8 pounds, 3.1 pounds, and 4.3 pounds, for patients receiving final doses of 30 mg, 50 mg, and 70 mg of lisdexamfetamine dimesylate, respectively, compared to a mean weight gain of 0.5 pounds for patients receiving placebo. Binge Eating Disorder The safety data in this section is based on data from two 12-week parallel group, flexible-dose, placebo-controlled studies in adults with BED [see Clinical Studies 14.2 ] . Patients with cardiovascular risk factors other than obesity and smoking were excluded. Adverse Reactions Associated with Discontinuation of Treatment in BED Clinical Trials In controlled trials of patients ages 18 to 55 years, 5.1% (19/373) of lisdexamfetamine dimesylate -treated patients discontinued due to adverse reactions compared to 2.4% (9/372) of placebo- treated patients. No single adverse reaction led to discontinuation in 1% or more of lisdexamfetamine dimesylate -treated patients. Less commonly reported adverse reactions (less than 1% or less than twice rate of placebo) included increased heart rate, headache, abdominal pain upper, dyspnea, rash, insomnia, irritability, feeling jittery and anxiety. Adverse Reactions Occurring at an Incidence of 5% or More and At Least Twice Placebo Among Lisdexamfetamine Dimesylate Treated Patients with BED in Clinical Trials The most common adverse reactions (incidence ≥5% and at a rate at least twice placebo) reported in adults were dry mouth, insomnia, decreased appetite, increased heart rate, constipation, feeling jittery, and anxiety. Adverse Reactions Occurring at an Incidence of 2% or More and At Least Twice Placebo Among Lisdexamfetamine Dimesylate Treated Patients with BED in Clinical Trials Adverse reactions reported in the pooled controlled trials in adult patients (Study 11 and 12) treated with lisdexamfetamine dimesylate or placebo are presented in Table 4 below.
Table 4 Adverse Reactions Reported by 2% or More of Adult Patients with BED Taking Lisdexamfetamine Dimesylate and Greater than or Equal to Twice the Incidence in Patients Taking Placebo in 12-Week Clinical Trials (Study 11 and 12)
| Lisdexamfetamine dimesylate (n=373) | Placebo (n=372) | |
| Dry Mouth | 36% | 7% |
| Insomnia• | 20% | 8% |
| Decreased Appetite | 8% | 2% |
| Increased Heart Rate† | 7% | 1% |
| Feeling Jittery | 6% | 1% |
| Constipation | 6% | 1% |
| Anxiety | 5% | 1% |
| Diarrhea | 4% | 2% |
| Decreased Weight | 4% | 0% |
| Hyperhidrosis | 4% | 0% |
| Vomiting | 2% | 1% |
| Gastroenteritis | 2% | 1% |
| Paresthesia | 2% | 1% |
| Pruritus | 2% | 1% |
| Upper Abdominal Pain | 2% | 0% |
| Energy Increased | 2% | 0% |
| Urinary Tract Infection | 2% | 0% |
| Nightmare | 2% | 0% |
| Restlessness | 2% | 0% |
| Oropharyngeal Pain | 2% | 0% |
•Includes all preferred terms containing the word "insomnia." † Includes the preferred terms "heart rate increased "and "tachycardia."
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of lisdexamfetamine dimesylate. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These events are as follows: cardiomyopathy, mydriasis, diplopia, difficulties with visual accommodation, blurred vision, eosinophilic hepatitis, anaphylactic reaction, hypersensitivity, dyskinesia, dysgeusia, motor and verbal tics, bruxism, depression, dermatillomania, alopecia, aggression, Stevens-Johnson Syndrome, chest pain, angioedema, urticaria, seizures, libido changes, frequent or prolonged erections, constipation, rhabdomyolysis, and intestinal ischemia.
DRUG INTERACTIONS
Acidifying and Alkalinizing Agents : Agents that alter urinary pH can alter blood levels of amphetamine. Acidifying agents decrease amphetamine blood levels, while alkalinizing agents increase amphetamine blood levels. Adjust ARYNTA dosage accordingly. (2.6 , 7.1 )
Drugs Having Clinically Important Interactions with Amphetamines
Table 5 Drugs Having Clinically Important Interactions with Amphetamines
| MAO Inhibitors (MAOI) | |
| Prevention or Management | Do not administer ARYNTA during or within 14 days following the administration of MAOI [see Contraindications (4 )] . |
| Mechanism and Clinical Effect(s) | MAOI antidepressants slow amphetamine metabolism, increasing amphetamines effect on the release of norepinephrine and other monoamines from adrenergic nerve endings causing headaches and other signs of hypertensive crisis. Toxic neurological effects and malignant hyperpyrexia can occur, sometimes with fatal results. |
| Serotonergic Drugs | |
| Prevention or Management | Initiate with lower doses and monitor patients for signs and symptoms of serotonin syndrome, particularly during ARYNTA initiation or dosage increase. If serotonin syndrome occurs, discontinue ARYNTA and the concomitant serotonergic drug(s) [see Warnings and Precautions (5.7 )] . |
| Mechanism and Clinical Effect(s) | The concomitant use of lisdexamfetamine dimesylate and serotonergic drugs increases the risk of serotonin syndrome. |
| CYP2D6 Inhibitors | |
| Prevention or Management | Initiate with lower doses and monitor patients for signs and symptoms of serotonin syndrome particularly during ARYNTA initiation and after a dosage increase. If serotonin syndrome occurs, discontinue ARYNTA and the CYP2D6 inhibitor [see Warnings and Precautions (5.7 ) and Overdosage (10 )]. |
| Mechanism and Clinical Effect(s) | The concomitant use of lisdexamfetamine dimesylate and CYP2D6 inhibitors may increase the exposure of dextroamphetamine, the active metabolite of lisdexamfetamine dimesylate compared to the use of the drug alone and increase the risk of serotonin syndrome. |
| Alkalinizing Agents | |
| Prevention or Management | Co-administration of ARYNTA and urinary alkalinizing agents should be avoided. |
| Mechanism and Clinical Effect(s) | Urinary alkalinizing agents can increase blood levels and potentiate the action of amphetamine. |
| Acidifying Agents | |
| Prevention or Management | Increase dose based on clinical response. |
| Mechanism and Clinical Effect(s) | Urinary acidifying agents can lower blood levels and efficacy of amphetamines. |
| Tricyclic Antidepressants | |
| Prevention or Management | Monitor frequently and adjust or use alternative therapy based on clinical response. |
| Mechanism and Clinical Effect(s) | May enhance the activity of tricyclic or sympathomimetic agents causing striking and sustained increases in the concentration of d-amphetamine in the brain; cardiovascular effects can be potentiated. |
DESCRIPTION
ARYNTA (lisdexamfetamine dimesylate), is a CNS stimulant. The chemical designation for lisdexamfetamine dimesylate is (2S)-2,6-diamino-N-[(1S)-1-methyl-2-phenylethyl] hexanamide dimethanesulfonate. The molecular formula is C 15 H 25 N 3 O∙(CH 4 O 3 S) 2 , which corresponds to a molecular weight of 455.60. The chemical structure is:
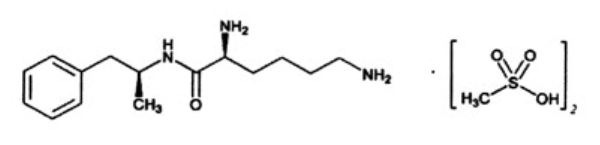
Lisdexamfetamine dimesylate is a white to off-white powder that is soluble in water (792 mg/mL). The pH is 4.24. The pKa values are 10.21 and 15.89. ARYNTA oral solution is a clear colorless solution, contains 10 mg/mL of lisdexamfetamine dimesylate (equivalent to 5.8 mg of lisdexamfetamine). Inactive ingredients: dibasic sodium phosphate dihydrate, methylparaben sodium, monobasic sodium phosphate dihydrate, propylparaben sodium, propylene glycol, saccharin sodium, hydrochloric acid, sodium hydroxide and purified water.
CLINICAL PHARMACOLOGY
Mechanism of Action
Lisdexamfetamine is a prodrug of dextroamphetamine. Amphetamines are non-catecholamine sympathomimetic amines with CNS stimulant activity. The exact mode of therapeutic action in ADHD and BED is not known.
Pharmacodynamics
Amphetamines block the reuptake of norepinephrine and dopamine into the presynaptic neuron and increase the release of these monoamines into the extraneuronal space. The parent drug, lisdexamfetamine, does not bind to the sites responsible for the reuptake of norepinephrine and dopamine in vitro.
Pharmacokinetics
No clinically significant difference in lisdexamfetamine and dextroamphetamine exposures was observed following oral administration of either ARYNTA or lisdexamfetamine dimesylate capsule. Pharmacokinetic studies after oral administration of lisdexamfetamine dimesylate have been conducted in healthy adult (capsule and chewable tablet formulations) and pediatric (6 to 12 years) patients with ADHD (capsule formulation). After single dose administration of lisdexamfetamine dimesylate, pharmacokinetics of dextroamphetamine was found to be linear between 30 mg and 70 mg in a pediatric study (6 to 12 years), and between 50 mg and 250 mg in an adult study. Dextroamphetamine pharmacokinetic parameters following administration of lisdexamfetamine dimesylate in adults exhibited low inter-subject (<25%) and intra-subject (<8%) variability. There is no accumulation of lisdexamfetamine and dextroamphetamine at steady state in healthy adults. Absorption Following single-dose administration of ARYNTA under fasted conditions, the peak plasma concentrations (Cmax) of lisdexamfetamine and dextroamphetamine were reached at approximately 1 hour and 3.5 hours post dose, respectively. Effect of Food No clinically significant differences in ARYNTA pharmacokinetics were observed following administration of a high-fat meal (1000 calories, 50% fat). Food delayed the median time to reach C max (T max ) of dextroamphetamine from 3.5 hours to 4.5 hours. Elimination Plasma concentrations of unconverted lisdexamfetamine are low and transient, generally becoming non-quantifiable by 8 hours after administration. The plasma elimination half-life of lisdexamfetamine typically averaged less than one hour in volunteers ages 6 years and older. The plasma elimination half-life of dextroamphetamine was approximately 8.6 to 9.5 hours in pediatric patients 6 to 12 years and 10 to 11.3 hours in healthy adults. Metabolism Lisdexamfetamine is converted to dextroamphetamine and l-lysine primarily in blood due to the hydrolytic activity of red blood cells after oral administration of lisdexamfetamine dimesylate. In vitro data demonstrated that red blood cells have a high capacity for metabolism of lisdexamfetamine; substantial hydrolysis occurred even at low hematocrit levels (33% of normal). Lisdexamfetamine is not metabolized by cytochrome P450 enzymes. Excretion Following oral administration of a 70 mg dose of radiolabeled lisdexamfetamine dimesylate to 6 healthy subjects, approximately 96% of the oral dose radioactivity was recovered in the urine and only 0.3% recovered in the feces over a period of 120 hours. Of the radioactivity recovered in the urine, 42% of the dose was related to amphetamine, 25% to hippuric acid, and 2% to intact lisdexamfetamine. Specific Populations Exposures of dextroamphetamine in specific populations are summarized in Figure 1.
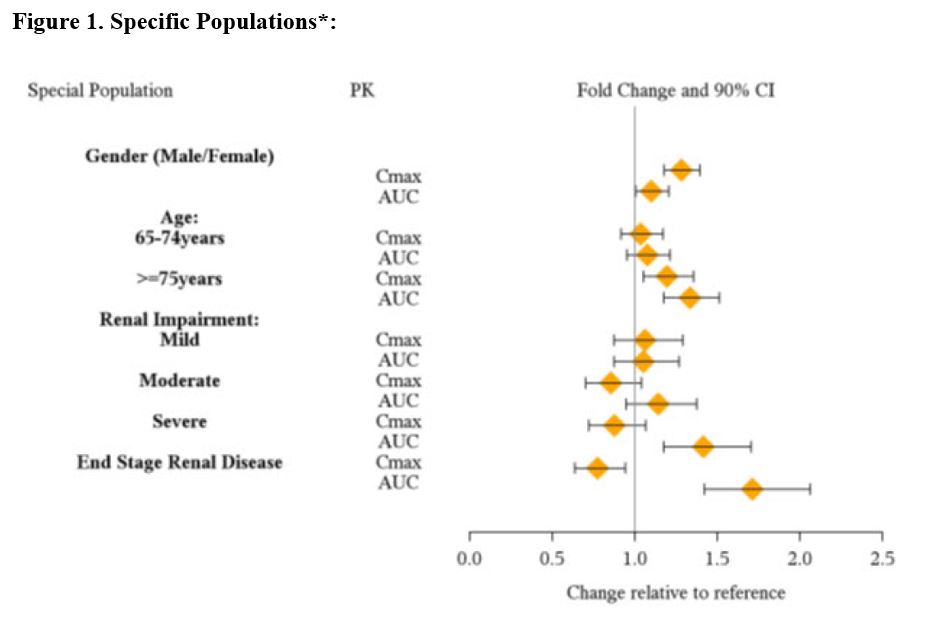
•Figure 1 shows the geometric mean ratios and the 90% confidence limits for C max and AUC of d-amphetamine. Comparison for gender uses males as the reference. Comparison for age uses 55-64 years as the reference.
Drug Interaction Studies
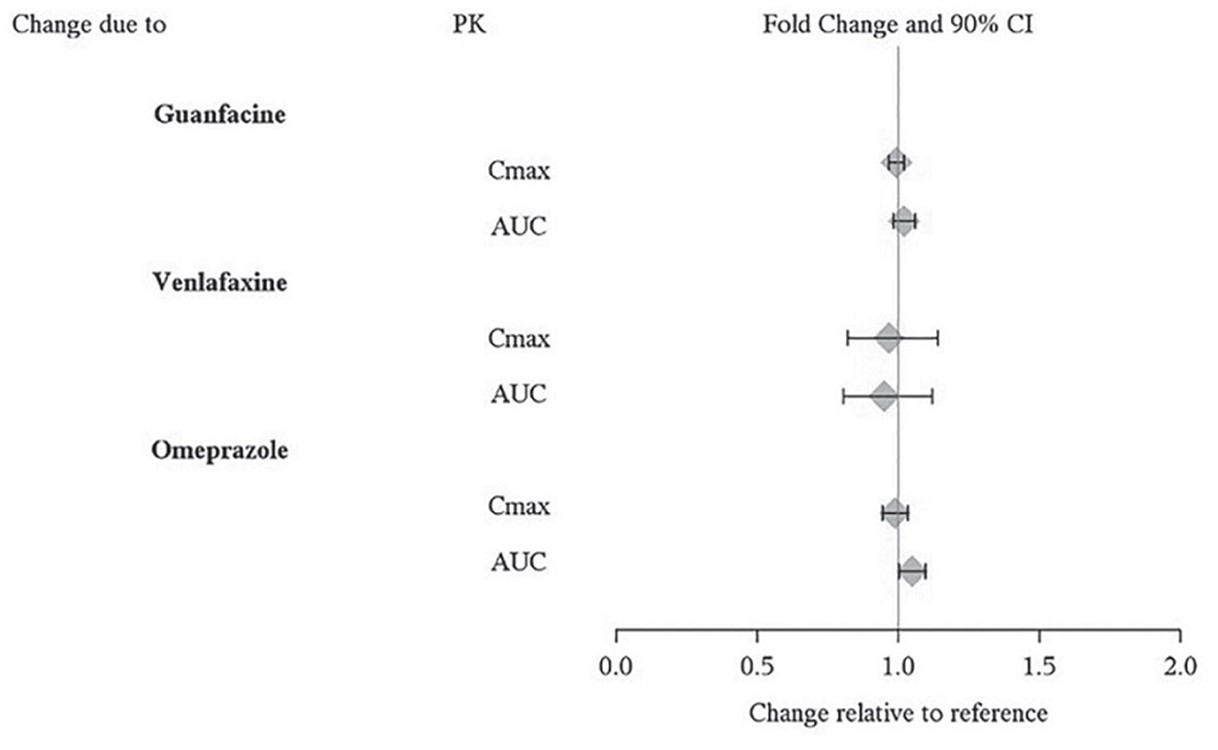
Effects of other drugs on the exposures of dextroamphetamine are summarized in Figure 2.
Figure 2. Effect of Other Drugs on Lisdexamfetamine Dimesylate
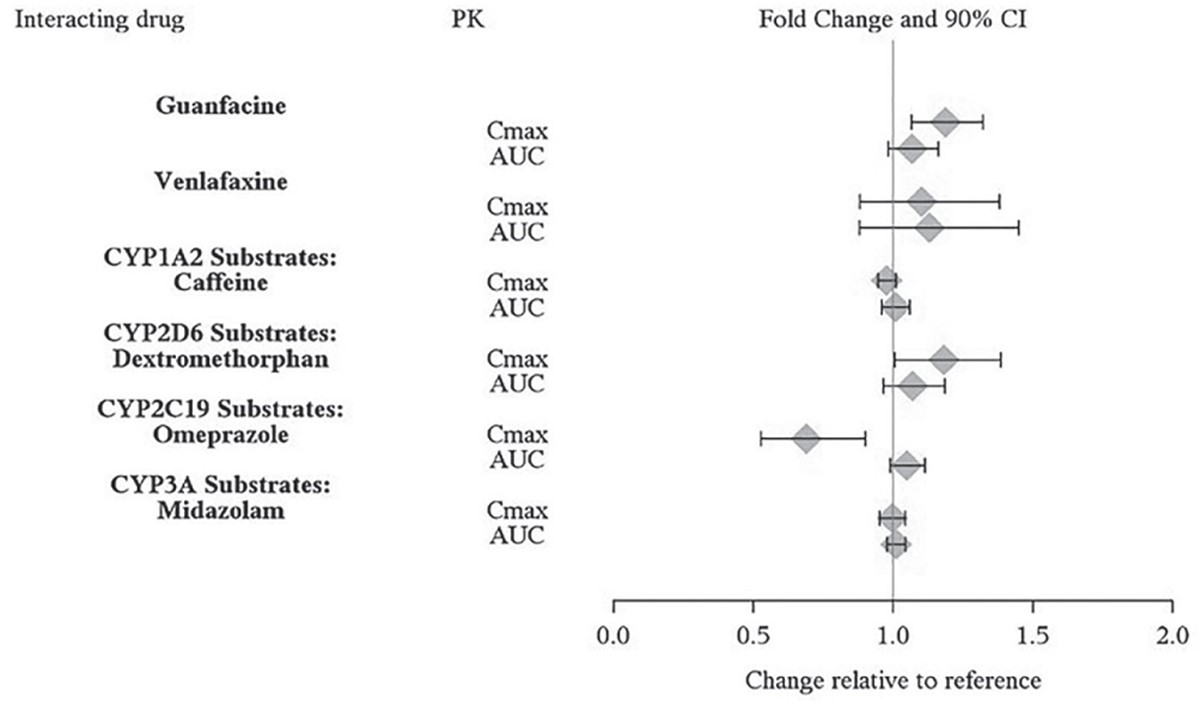
The effects of lisdexamfetamine dimesylate on the exposures of other drugs are summarized in Figure 3.
Figure 3. Effect of Lisdexamfetamine Dimesylate on Other Drugs
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenesis Carcinogenicity studies of lisdexamfetamine dimesylate have not been performed. No evidence of carcinogenicity was found in studies in which d-, l-amphetamine (enantiomer ratio of 1:1) was administered to mice and rats in the diet for 2 years at doses of up to 30 mg/kg/day in male mice, 19 mg/kg/day in female mice, and 5 mg/kg/day in male and female rats. Mutagenesis Lisdexamfetamine dimesylate was not clastogenic in the mouse bone marrow micronucleus test in vivo and was negative when tested in the E. coli and S. typhimurium components of the Ames test and in the L5178Y/TK+/- mouse lymphoma assay in vitro. Impairment of Fertility Amphetamine (d- to l-enantiomer ratio of 3:1) did not adversely affect fertility or early embryonic development in the rat at doses of up to 20 mg/kg/day.
Animal Toxicology and/or Pharmacology
Acute administration of high doses of amphetamine (d- or d, l-) has been shown to produce long-lasting neurotoxic effects, including irreversible nerve fiber damage, in rodents. The significance of these findings to humans is unknown.
CLINICAL STUDIES
The efficacy of ARYNTA has been established based on adequate and well-controlled studies of oral lisdexamfetamine dimesylate in the treatment of adults and pediatric patients 6 years and older with ADHD and adults with moderate to severe binge eating disorder (BED). The information below describes the results of the adequate and well-controlled studies of oral lisdexamfetamine dimesylate in patients with ADHD and BED.
Attention Deficit Hyperactivity Disorder (ADHD)
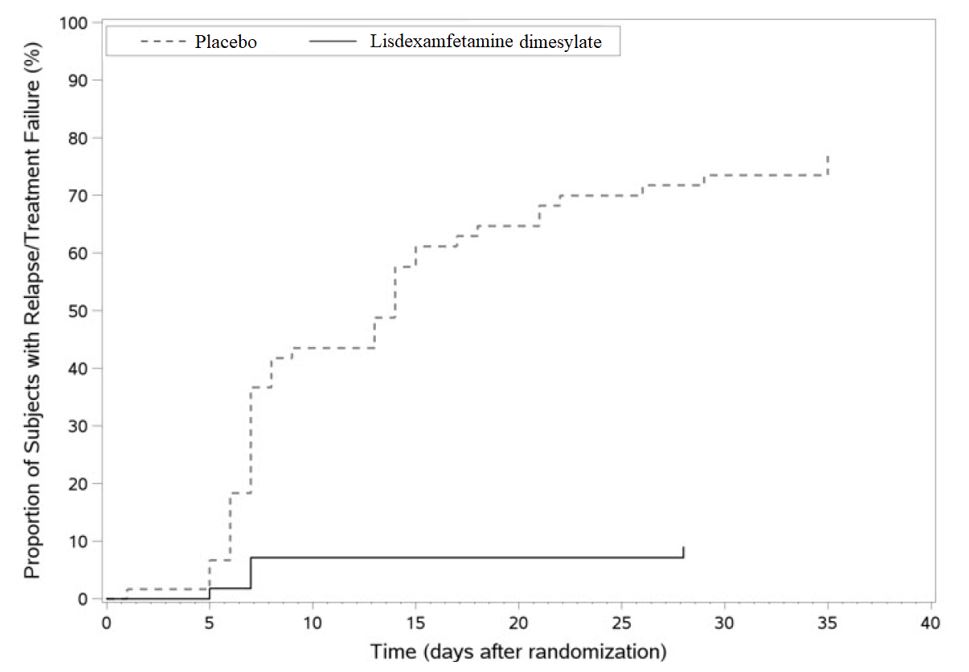
Pediatric Patients Ages 6 to 12 Years with ADHD A double-blind, randomized, placebo-controlled, parallel-group study (Study 1) was conducted in pediatric patients ages 6 to 12 years (N=290) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Patients were randomized to receive final doses of 30 mg, 50 mg, or 70 mg of lisdexamfetamine dimesylate or placebo once daily in the morning for a total of four weeks of treatment. All patients receiving lisdexamfetamine dimesylate were initiated on 30 mg for the first week of treatment. Patients assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS), an 18-item questionnaire with a score range of 0-54 points that measures the core symptoms of ADHD which includes both hyperactive/impulsive and inattentive subscales. Endpoint was defined as the last post-randomization treatment week (i.e., Weeks 1 through 4) for which a valid score was obtained. All lisdexamfetamine dimesylate dose groups were superior to placebo in the primary efficacy outcome. Mean effects at all doses were similar; however, the highest dose (70 mg/day) was numerically superior to both lower doses (Study 1 in Table 6). The effects were maintained throughout the day based on parent ratings (Conners' Parent Rating Scale) in the morning (approximately 10 am), afternoon (approximately 2 pm), and early evening (approximately 6 pm). A double-blind, placebo-controlled, randomized, crossover design, analog classroom study (Study 2) was conducted in pediatric patients ages 6 to 12 years (N=52) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Following a 3-week open-label dose optimization with Adderall XR ® , patients were randomly assigned to continue their optimized dose of Adderall XR (10 mg, 20 mg, or 30 mg), lisdexamfetamine dimesylate (30 mg, 50 mg, or 70 mg), or placebo once daily in the morning for 1 week each treatment. Efficacy assessments were conducted at 1, 2, 3, 4.5, 6, 8, 10, and 12 hours post-dose using the Swanson, Kotkin, Agler, M.Flynn, and Pelham Deportment scores (SKAMP-DS), a 4-item subscale of the SKAMP with scores ranging from 0 to 24 points that measures deportment problems leading to classroom disruptions. A significant difference in patient behavior, based upon the average of investigator ratings on the SKAMP-DS across the 8 assessments were observed between patients when they received lisdexamfetamine dimesylate compared to patients when they received placebo (Study 2 in Table 6). The drug effect reached statistical significance from hours 2 to 12 post-dose, but was not significant at 1 hour. A second double-blind, placebo-controlled, randomized, crossover design, analog classroom study (Study 3) was conducted in pediatric patients ages 6 to 12 years (N=129) who met DSM-IV criteria for ADHD (either the combined type or the hyperactive-impulsive type). Following a 4-week open-label dose optimization with lisdexamfetamine dimesylate (30 mg, 50 mg, 70 mg), patients were randomly assigned to continue their optimized dose of lisdexamfetamine dimesylate or placebo once daily in the morning for 1 week each treatment. A significant difference in patient behavior, based upon the average of investigator ratings on the SKAMP-Deportment scores across all 7 assessments conducted at 1.5, 2.5, 5.0, 7.5, 10.0, 12.0, and 13.0 hours post-dose, were observed between patients when they received lisdexamfetamine dimesylate compared to patients when they received placebo (Study 3 in Table 6, Figure 4). Pediatric Patients Ages 13 to 17 Years with ADHD A double-blind, randomized, placebo-controlled, parallel-group study (Study 4) was conducted in pediatric patients ages 13 to 17 years (N=314) who met DSM-IV criteria for ADHD. In this study, patients were randomized in a 1:1:1:1 ratio to a daily morning dose of lisdexamfetamine dimesylate (30 mg/day, 50 mg/day or 70 mg/day) or placebo for a total of four weeks of treatment. All patients receiving lisdexamfetamine dimesylate were initiated on 30 mg for the first week of treatment. Patients assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS). Endpoint was defined as the last post-randomization treatment week (i.e., Weeks 1 through 4) for which a valid score was obtained. All lisdexamfetamine dimesylate dose groups were superior to placebo in the primary efficacy outcome (Study 4 in Table 6). Pediatric Patients Ages 6 to 17 Years: Short-Term Treatment in ADHD A double-blind, randomized, placebo- and active-controlled parallel-group, dose-optimization study (Study 5) was conducted in pediatric patients ages 6 to 17 years (n=336) who met DSM-IV criteria for ADHD. In this eight-week study, patients were randomized to a daily morning dose of lisdexamfetamine dimesylate (30, 50 or 70 mg/day), an active control, or placebo (1:1:1). The study consisted of a Screening and Washout Period (up to 42 days), a 7-week Double-blind Evaluation Period (consisting of a 4-week Dose-Optimization Period followed by a 3-week Dose-Maintenance Period), and a 1-week Washout and Follow-up Period. During the Dose Optimization Period, subjects were titrated until an optimal dose, based on tolerability and investigator's judgment, was reached. lisdexamfetamine dimesylate showed significantly greater efficacy than placebo. The placebo-adjusted mean reduction from baseline in the ADHD-RS-IV total score was 18.6. Subjects on lisdexamfetamine dimesylate also showed greater improvement on the Clinical Global Impression-Improvement (CGI-I) rating scale compared to subjects on placebo (Study 5 in Table 6). Pediatric Patients Ages 6 to 17 Years: Maintenance Treatment in ADHD Maintenance of Efficacy Study (Study 6) – A double-blind, placebo-controlled, randomized withdrawal study was conducted in pediatric patients ages 6 to 17 years (N=276) who met the diagnosis of ADHD (DSM-IV criteria). A total of 276 patients were enrolled into the study, 236 patients participated in Study 5 and 40 subjects directly enrolled. Subjects were treated with open-label lisdexamfetamine dimesylate for at least 26 weeks prior to being assessed for entry into the randomized withdrawal period. Eligible patients had to demonstrate treatment response as defined by CGI-S <3 and Total Score on the ADHD-RS ≤22. Patients that maintained treatment response for 2 weeks at the end of the open label treatment period were eligible to be randomized to ongoing treatment with the same dose of lisdexamfetamine dimesylate (N=78) or switched to placebo (N=79) during the double-blind phase. Patients were observed for relapse (treatment failure) during the 6 week double-blind phase. A significantly lower proportion of treatment failures occurred among lisdexamfetamine dimesylate subjects (15.8%) compared to placebo (67.5%) at endpoint of the randomized withdrawal period. The endpoint measurement was defined as the last post-randomization treatment week at which a valid ADHD-RS Total Score and CGI-S were observed. Treatment failure was defined as a ≥50% increase (worsening) in the ADHD-RS Total Score and a ≥2-point increase in the CGI-S score compared to scores at entry into the double-blind randomized withdrawal phase. Subjects who withdrew from the randomized withdrawal period and who did not provide efficacy data at their last on-treatment visit were classified as treatment failures (Study 6, Figure 5). Adults: Short-Term Treatment in ADHD A double-blind, randomized, placebo-controlled, parallel-group study (Study 7) was conducted in adults ages 18 to 55 (N=420) who met DSM-IV criteria for ADHD. In this study, patients were randomized to receive final doses of 30 mg, 50 mg, or 70 mg of lisdexamfetamine dimesylate or placebo for a total of four weeks of treatment. All patients receiving lisdexamfetamine dimesylate were initiated on 30 mg for the first week of treatment. Patients assigned to the 50 mg and 70 mg dose groups were titrated by 20 mg per week until they achieved their assigned dose. The primary efficacy outcome was change in Total Score from baseline to endpoint in investigator ratings on the ADHD Rating Scale (ADHD-RS). Endpoint was defined as the last post-randomization treatment week (i.e., Weeks 1 through 4) for which a valid score was obtained. All lisdexamfetamine dimesylate dose groups were superior to placebo in the primary efficacy outcome (Study 7 in Table 6). The second study was a multi-center, randomized, double-blind, placebo-controlled, cross-over, modified analog classroom study (Study 8) of lisdexamfetamine dimesylate to simulate a workplace environment in 142 adults ages 18 to 55 who met DSM-IV-TR criteria for ADHD. There was a 4-week open-label, dose optimization phase with lisdexamfetamine dimesylate (30 mg/day, 50 mg/day, or 70 mg/day in the morning). Patients were then randomized to one of two treatment sequences: 1) lisdexamfetamine dimesylate (optimized dose) followed by placebo, each for one week, or 2) placebo followed by lisdexamfetamine dimesylate, each for one week. Efficacy assessments occurred at the end of each week, using the Permanent Product Measure of Performance (PERMP), a skill-adjusted math test that measures attention in ADHD. PERMP total score results from the sum of the number of math problems attempted plus the number of math problems answered correctly. lisdexamfetamine dimesylate treatment, compared to placebo, resulted in a statistically significant improvement in attention across all post-dose time points, as measured by average PERMP total scores over the course of one assessment day, as well as at each time point measured. The PERMP assessments were administered at pre-dose (-0.5 hours) and at 2, 4, 8, 10, 12, and 14 hours post-dose (Study 8 in Table 6, Figure 6). Adults: Maintenance Treatment in ADHD A double-blind, placebo-controlled, randomized withdrawal design study (Study 9) was conducted in adults ages 18 to 55 (N=123) who had a documented diagnosis of ADHD or met DSM-IV criteria for ADHD. At study entry, patients must have had documentation of treatment with lisdexamfetamine dimesylate for a minimum of 6 months and had to demonstrate treatment response as defined by Clinical Global Impression Severity (CGI-S) ≤3 and Total Score on the ADHD-RS <22. ADHD-RS Total Score is a measure of core symptoms of ADHD. The CGI-S score assesses the clinician's impression of the patient's current illness state and ranges from 1 (not at all ill) to 7 (extremely ill). Patients that maintained treatment response at Week 3 of the open label treatment phase (N=116) were eligible to be randomized to ongoing treatment with the same dose of lisdexamfetamine dimesylate (N=56) or switched to placebo (N=60) during the double-blind phase. Patients were observed for relapse (treatment failure) during the 6-week double-blind phase. The efficacy endpoint was the proportion of patients with treatment failure during the double-blind phase. Treatment failure was defined as a ≥50% increase (worsening) in the ADHD-RS Total Score and ≥2-point increase in the CGI-S score compared to scores at entry into the double-blind phase. Maintenance of efficacy for patients treated with lisdexamfetamine dimesylate was demonstrated by the significantly lower proportion of patients with treatment failure (9%) compared to patients receiving placebo (75%) at endpoint during the double-blind phase (Study 9, Figure 7).
Table 6: Summary of Primary Efficacy Results from Short-term Studies of Lisdexamfetamine dimesylate in Pediatric Patients (Ages 6 to 17) and Adults with ADHD
| Study Number (Age range) | Primary Endpoint | Treatment Group | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference (95% CI) |
| Study 1 (6 – 12 years) | ADHD-RS-IV | Lisdexamfetamine dimesylate (30 mg/day) † | 43.2 (6.7) | -21.8 (1.6) | -15.6 (-19.9, -11.2) |
| Lisdexamfetamine dimesylate (50 mg/day) † | 43.3 (6.7) | -23.4 (1.6) | -17.2 (-21.5, -12.9) | ||
| Lisdexamfetamine dimesylate (70 mg/day) † | 45.1(6.8) | -26.7 (1.5) | -20.5 (-24.8, -16.2) | ||
| Placebo | 42.4 (7.1) | -6.2 (1.6) | -- | ||
| Study 2 (6 – 12 years) | Average SKAMP-DS | Lisdexamfetamine dimesylate (30, 50 or 70 mg/day) † | -- ‡ | 0.8 (0.1) § | -0.9 (-1.1, -0.7) |
| Placebo | -- ‡ | 1.7 (0.1) § | -- | ||
| Study 3 (6 – 12 years) | Average SKAMP-DS | Lisdexamfetamine dimesylate (30, 50 or 70 mg/day) † | 0.9 (1.0) ¶ | 0.7 (0.1) | -0.7 (-0.9, -0.6) |
| Placebo | 0.7 (0.9) ¶ | 1.4 (0.1) | -- | ||
| Study 4 (13 – 17 years) | ADHD-RSIV | Lisdexamfetamine dimesylate (30 mg/day) † | 38.3 (6.7) | -18.3 (1.2) | -5.5 (-9.0, -2.0) |
| Lisdexamfetamine dimesylate (50 mg/day) † | 37.3 (6.3) | -21.1 (1.3) | -8.3 (-11.8, -4.8) | ||
| Lisdexamfetamine dimesylate (70 mg/day) † | 37.0 (7.3) | -20.7 (1.3) | -7.9 (-11.4, -4.5) | ||
| Placebo | 38.5 (7.1) | -12.8 (1.2) | -- | ||
| Study 5 (6 – 17 years) | ADHD-RS-IV | Lisdexamfetamine dimesylate (30, 50 or 70 mg/day) † | 40.7 (7.3) | -24.3 (1.2) | -18.6 (-21.5, -15.7) |
| Placebo | 41.0 (7.1) | -5.7 (1.1) | -- | ||
| Study 7 (18 – 55 years) | ADHD-RS-IV | Lisdexamfetamine dimesylate (30 mg/day) † | 40.5 (6.2) | -16.2 (1.1) | -8.0 (-11.5, -4.6) |
| Lisdexamfetamine dimesylate (50 mg/day) † | 40.8 (7.3) | -17.4 (1.0) | -9.2 (-12.6, -5.7) | ||
| Lisdexamfetamine dimesylate (70 mg/day) † | 41.0 (6.0) | -18.6 (1.0) | -10.4 (-13.9, -6.9) | ||
| Placebo | 39.4 (6.4) | -8.2 (1.4) | -- | ||
| Study 8 (18 – 55 years) | Average PERMP | Lisdexamfetamine dimesylate (30, 50 or 70 mg/day) † | 260.1 (86.2) ¶ | 312.9 (8.6) § | 23.4 (15.6, 31.2) |
| Placebo | 261.4 (75.0) ¶ | 289.5 (8.6) § | -- |
SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval. • Difference (drug minus placebo) in least-squares mean change from baseline.
† Doses statistically significantly superior to placebo. ‡ Pre-dose SKAMP-DS was not collected. § LS Mean for SKAMP-DS (Study 2 and 3) or PERMP (Study 8) is post-dose average score over all sessions of the treatment day, rather than change from baseline. ¶ Pre-dose SKAMP-DS (Study 3) or PERMP (Study 8) total score, averaged over both periods.
Figure 4 LS Mean SKAMP Deportment Subscale Score by Treatment and Time-point for Pediatric Patients Ages 6 to 12 with ADHD after 1 Week of Double-Blind Treatment (Study 3)
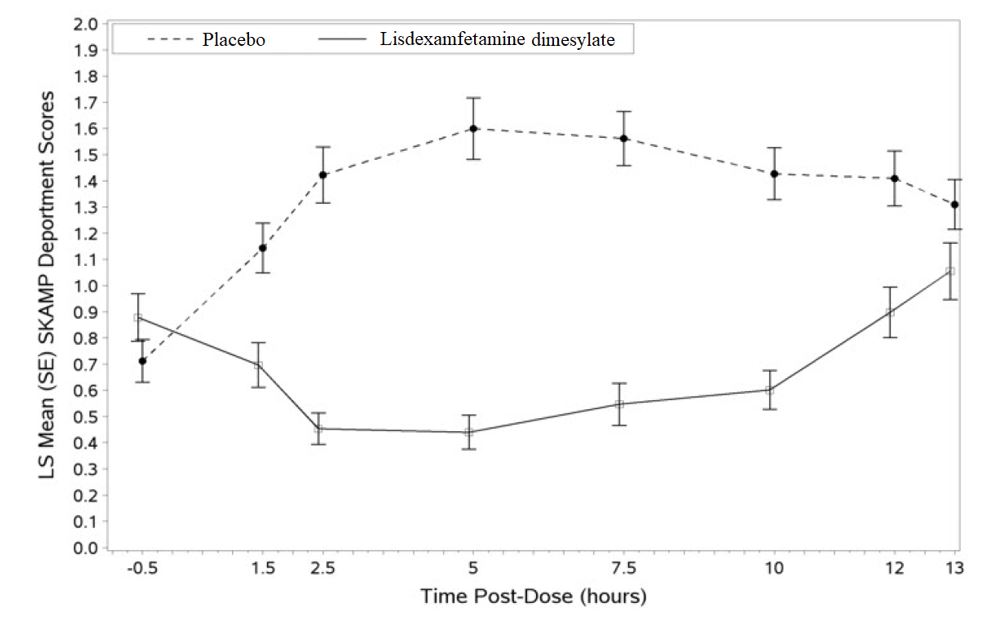
Higher score on the SKAMP-Deportment scale indicates more severe symptoms.
Figure 5 Kaplan-Meier Estimated Proportion of Patients with Treatment Failure for Pediatric Patients Ages 6 to 17 (Study 6)
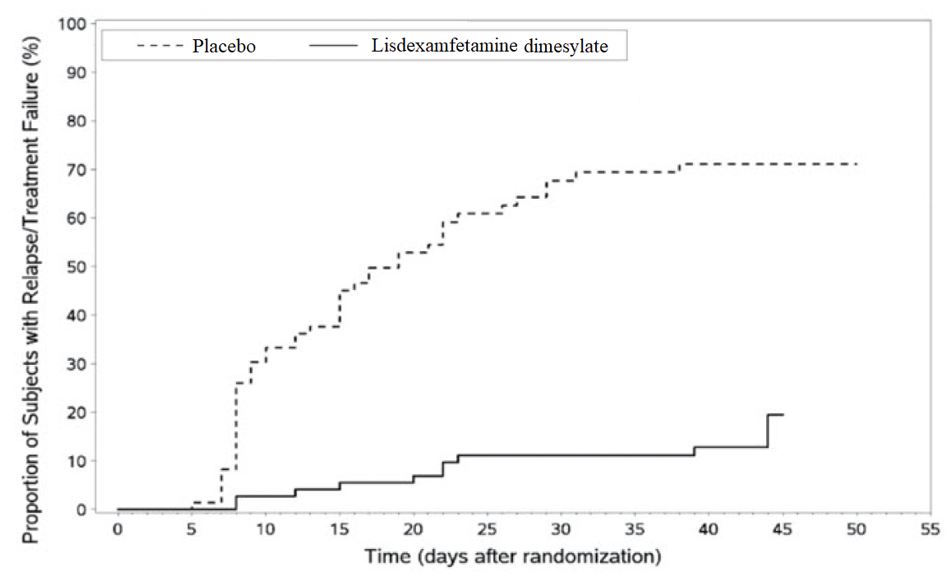
Figure 6 LS Mean (SE) PERMP Total Score by Treatment and Time-point for Adults Ages 18 to 55 with ADHD after 1 Week of Double-Blind Treatment (Study 8)
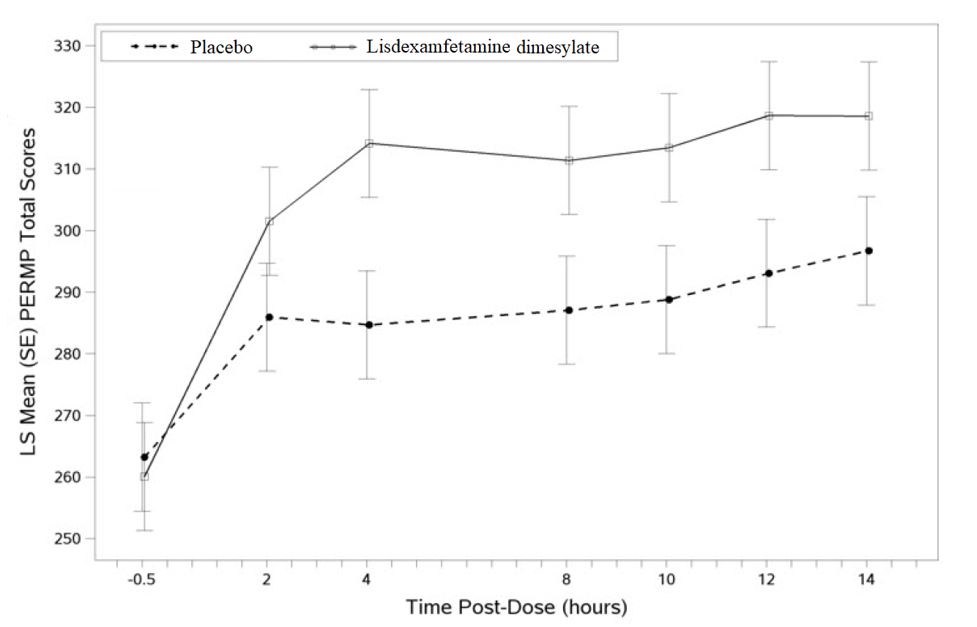
Higher score on the PERMP scale indicates less severe symptoms.
Figure 7 Kaplan-Meier Estimated Proportion of Subjects with Relapse in Adults with ADHD (Study 9)
Binge Eating Disorder (BED)
A phase 2 study evaluated the efficacy of lisdexamfetamine dimesylate 30, 50 and 70 mg/day compared to placebo in reducing the number of binge days/week in adults with at least moderate to severe BED. This randomized, double-blind, parallel-group, placebo-controlled, forced-dose titration study (Study 10) consisted of an 11-week double-blind treatment period (3 weeks of forced-dose titration followed by 8 weeks of dose maintenance). Lisdexamfetamine dimesylate 30 mg/day was not statistically different from placebo on the primary endpoint. The 50 and 70 mg/day doses were statistically superior to placebo on the primary endpoint. The efficacy of lisdexamfetamine dimesylate in the treatment of BED was demonstrated in two 12-week randomized, double-blind, multi-center, parallel-group, placebo-controlled, dose-optimization studies (Study 11 and Study 12) in adults aged 18-55 years (Study 11: N=374, Study 12: N=350) with moderate to severe BED. A diagnosis of BED was confirmed using DSM-IV criteria for BED. Severity of BED was determined based on having at least 3 binge days per week for 2 weeks prior to the baseline visit and on having a Clinical Global Impression Severity (CGI-S) score of ≥4 at the baseline visit. For both studies, a binge day was defined as a day with at least 1 binge episode, as determined from the subject's daily binge diary. Both 12-week studies consisted of a 4-week dose-optimization period and an 8-week dose-maintenance period. During dose-optimization, subjects assigned to lisdexamfetamine dimesylate began treatment at the titration dose of 30 mg/day and, after 1 week of treatment, were subsequently titrated to 50 mg/day. Additional increases to 70 mg/day were made as tolerated and clinically indicated. Following the dose-optimization period, subjects continued on their optimized dose for the duration of the dose-maintenance period. The primary efficacy outcome for the two studies was defined as the change from baseline at Week 12 in the number of binge days per week. Baseline is defined as the weekly average of the number of binge days per week for the 14 days prior to the baseline visit. Subjects from both studies on lisdexamfetamine dimesylate had a statistically significantly greater reduction from baseline in mean number of binge days per week at Week 12. In addition, subjects on lisdexamfetamine dimesylate showed greater improvement as compared to placebo across key secondary outcomes with higher proportion of subjects rated improved on the CGI-I rating scale, higher proportion of subjects with 4-week binge cessation, and greater reduction in the Yale-Brown Obsessive Compulsive Scale Modified for Binge Eating (Y-BOCS-BE) total score.
Table 7: Summary of Primary Efficacy Results in BED
| Study Number | Treatment Group | Primary Efficacy Measure: Binge Days per Week at Week 12 | ||
| Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference• (95% CI) | ||
| Study 11 | Lisdexamfetamine dimesylate (50 mg or 70 mg/day)† | 4.79 (1.27) | -3.87 (0.12) | -1.35 (-1.70, -1.01) |
| Placebo | 4.60 (1.21) | -2.51 (0.13) | -- | |
| Study 12 | Lisdexamfetamine dimesylate (50 mg or 70 mg/day)† | 4.66 (1.27) | -3.92 (0.14) | -1.66 (-2.04, -1.28) |
| Placebo | 4.82 (1.42) | -2.26 (0.14) | -- | |
SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval. • Difference (drug minus placebo) in least-squares mean change from baseline. † Doses statistically significantly superior to placebo. A double-blind, placebo controlled, randomized withdrawal design study (Study 13) was conducted to evaluate maintenance of efficacy based on time to relapse between lisdexamfetamine dimesylate and placebo in adults aged 18 to 55 (N=267) with moderate to severe BED. In this longer-term study patients who had responded to lisdexamfetamine dimesylate in the preceding 12-week open-label treatment phase were randomized to continuation of lisdexamfetamine dimesylate or placebo for up to 26 weeks of observation for relapse. Response in the open-label phase was defined as 1 or fewer binge days each week for four consecutive weeks prior to the last visit at the end of the 12-week open-label phase and a CGI-S score of 2 or less at the same visit. Relapse during the double-blind phase was defined as having 2 or more binge days each week for two consecutive weeks (14 days) prior to any visit and having an increase in CGI-S score of 2 or more points compared to the randomized-withdrawal baseline. Maintenance of efficacy for patients who had an initial response during the open-label period and then continued on lisdexamfetamine dimesylate during the 26-week double-blind randomized-withdrawal phase was demonstrated with lisdexamfetamine dimesylate being superior over placebo as measured by time to relapse. Figure 8 Kaplan-Meier Estimated Proportions of Subjects with Relapse in Adults with BED (Study 13)
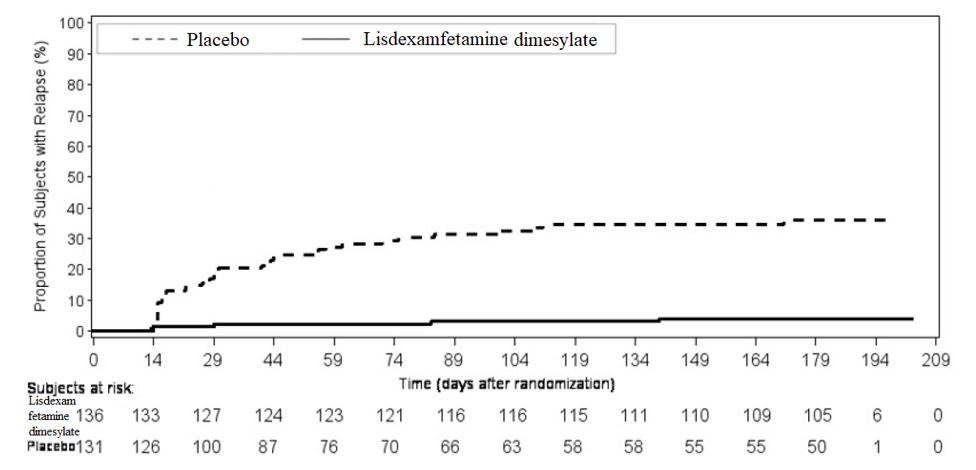
Examination of population subgroups based on age (there were no patients over 65), gender, and race did not reveal any clear evidence of differential responsiveness in the treatment of BED.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ARYNTA oral solution is a clear colorless solution contains 10 mg/mL of lisdexamfetamine dimesylate. It is packaged in 100 mL PET bottles with a child resistant closure and in 30 mL, 60 mL, 90 mL, 120 mL HDPE bottles with child resistant closures as follows: Each sealed bottle is co-packaged with one press-in bottle adapter and one oral dosing syringe.
| NDC Number | Size |
| 24338-019-01 | 100 mL |
| 24338-103-01 | 30 mL |
| 24338-160-01 | 60 mL |
| 24338-109-01 | 90 mL |
| 24338-162-01 | 120 mL |
Storage and Handling
ARYNTA is stored at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Keep the container tightly closed. Throw away (discard) any remaining ARYNTA 30 days after first opening the bottle.
Mechanism of Action
Lisdexamfetamine is a prodrug of dextroamphetamine. Amphetamines are non-catecholamine sympathomimetic amines with CNS stimulant activity. The exact mode of therapeutic action in ADHD and BED is not known.