Get your patient on Aqvesme (Mitapivat)
Aqvesme prescribing information
WARNING: HEPATOCELLULAR INJURY
AQVESME can cause serious hepatocellular injury. Measure liver laboratory tests (ALT, AST, alkaline phosphatase, and total bilirubin with fractionation) at baseline and every 4 weeks for 24 weeks and then as clinically indicated. Avoid use of AQVESME in patients with cirrhosis. Discontinue AQVESME if hepatic injury is suspected [see Warnings and Precautions (5.1 )] .
Because of the risk of hepatocellular injury, AQVESME is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the AQVESME REMS [see Warnings and Precautions (5.2 )] .
INDICATIONS AND USAGE
AQVESME is indicated for the treatment of anemia in adults with alpha- or beta-thalassemia.
DOSAGE AND ADMINISTRATION
2.1 Important Dosage and Administration Information
- AQVESME is taken with or without food.
- Swallow tablets whole. Do not split, crush, chew, or dissolve the tablets.
- If a dose of AQVESME is missed by 4 hours or less, administer the dose as soon as possible. If a dose of AQVESME is missed by more than 4 hours, do not administer a replacement dose, and wait until the next scheduled dose. Subsequently, return to the normal dosing schedule.
- Monitor for hepatocellular injury during treatment with AQVESME [see Dosage and Administration (2.3) ] .
2.2 Recommended Dosage
The recommended dosage for adults with alpha- or beta-thalassemia is AQVESME 100 mg orally twice daily.
Treatment with AQVESME is intended to be long-term. Discontinue AQVESME if no benefit in hemolytic anemia has been observed, based on the totality of laboratory results and clinical status of the patient, unless there is another explanation for response failure (e.g., bleeding, surgery, other concomitant illnesses).
Interruption or Discontinuation
If a patient needs to interrupt or discontinue AQVESME for any reason, a dose taper is not necessary.
2.3 Monitoring for Safety
Prior to Initiating Treatment with AQVESME
- Check liver tests including ALT, AST, alkaline phosphatase, total bilirubin with fractionation, before first AQVESME dose.
During Treatment with AQVESME
- After the first dose, check liver tests including ALT, AST, alkaline phosphatase, total bilirubin with fractionation every 4 weeks for 24 weeks and as clinically indicated thereafter.
When Drug-Induced Liver Injury Is Suspected
- Interrupt AQVESME and complete a comprehensive evaluation to rule out other causes of liver injury.
- If AQVESME-related liver injury caused new or worsening jaundice or ALT ≥10×baseline, do NOT restart AQVESME.
- If AQVESME-related liver injury is not ruled out, but peak ALT is <10×baseline without elevation of bilirubin above baseline, and if AQVESME is resumed, reinitiate liver test monitoring every 4 weeks for 24 additional weeks.
- If AQVESME-related liver injury is ruled out, AQVESME may be restarted at provider discretion. Resume liver test monitoring schedule that existed prior to stopping AQVESME.
AQVESME Interruption Due to Non-Liver Causes
- If AQVESME was stopped for any reason for ≤8 weeks other than suspected AQVESME-related liver injury, resume the liver test monitoring schedule that existed prior to stopping AQVESME.
- If AQVESME was stopped for more than 8 weeks, restart liver test monitoring every 4 weeks for 24 additional weeks upon resumption of treatment with AQVESME.
- If treatment is stopped for any duration after 24 weeks of monitoring and treatment, resume monitoring as clinically indicated.
2.4 Recommended Dosage for Drug Interactions
Moderate CYP3A Inducers
Consider alternative therapies that are not moderate CYP3A inducers during treatment with AQVESME. If there are no alternative therapies, monitor Hb and do not exceed the maximum recommended dose of 100 mg orally twice daily [see Drug Interactions (7.1) and Clinical Pharmacology (12.3) ] .
DOSAGE FORMS AND STRENGTHS
- 100 mg tablets: oblong, blue, film-coated tablets with "M100" printed on both sides.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Available data from clinical trials of AQVESME are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes.
In animal reproduction studies, mitapivat orally administered twice daily to pregnant rats and rabbits during organogenesis was not teratogenic at exposures up to 9.9- and 2.4‑fold the human exposure associated with the MRHD, respectively. Mitapivat administered orally to pregnant rats twice daily during organogenesis through lactation did not result in adverse developmental effects at doses up to 9.9 times the MRHD ( see Data) .
The estimated background risk of major birth defects for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal Risk
Transfusion requirements in thalassemia patients are increased during pregnancy. Pregnant women with transfusion-dependent thalassemia are considered high risk, with their major complications being cardiac in origin, including cardiac dysrhythmia, right ventricular dysfunction, and cardiac failure, reported in 1.1% to 15.6%. Pregnant women with non-transfusion-dependent thalassemia need to be monitored and treated for risk of thrombosis, especially those who are splenectomized or infrequently transfused. Patients with non-transfusion-dependent thalassemia may develop a need for regular transfusions during pregnancy, and in such patients the risk of alloimmunization should be carefully evaluated. All pregnant women with thalassemia should be closely monitored for iron overload.
Data
Animal Data
In an embryo-fetal development study in rats, mitapivat was administered at doses of 5, 10, 25, and 100 mg/kg twice daily by oral gavage during the period of organogenesis (gestation days 6 to 17). There was a statistically significant 14% decrease in maternal net body weight gain at 100 mg/kg twice daily with associated decrease in food consumption. Enlarged or fused placenta and/or a distended amniotic sac, an increase in post-implantation loss (early and late resorptions), a decrease in the mean number of viable fetuses, lower mean fetal weights, and fetal external, visceral, and skeletal malformations were observed at 100 mg/kg twice daily, (48 times the MRHD, based on area under the plasma drug concentration-time curve [AUC]). No maternal or embryo-fetal toxicity was observed up to 25 mg/kg twice daily (9.9 times the MRHD, based on AUC).
In an embryo-fetal development study in rabbits, mitapivat was administered at doses of 12.5, 30, and 62.5 mg/kg twice daily by oral gavage during the period of organogenesis (gestation days 7 to 20). Lower fetal weight was observed at 62.5 mg/kg twice daily (2.4 times MRHD, based on AUC) and correlated with reduced maternal body weight gain. No effects on fetal morphology were observed.
In a pre- and post-natal development study in rats, mitapivat was administered at doses of 5, 10, 25, and 100 mg/kg twice daily by oral gavage during the period of organogenesis and continuing to weaning (gestation day 7 to lactation day 20). Dystocia was observed at ≥25 mg/kg twice daily (≥9.9 times MRHD, based on AUC). At 100 mg/kg twice daily (48 times MRHD, based on AUC) decreased maternal body weight gain, prolonged parturition, and dystocia occurred and resulted in maternal mortality, complete litter loss, decreased pup viability and decreased pup body weight. No adverse effects on pup growth and development, and reproductive performance were observed up to 50 mg/kg (9.9 times the MRHD, based on AUC).
Lactation
Risk Summary
There are no data on the presence of AQVESME or its metabolites in human or animal milk, the effects on the breastfed child, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for AQVESME and any potential adverse effects on the breastfed child from AQVESME or from the underlying maternal condition.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Clinical studies of AQVESME did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from younger subjects.
8.6 Hepatic Impairment
Avoid use in patients with cirrhosis (Child-Pugh Class A, B, or C) due to an increased risk of severe outcomes if hepatocellular injury occurs [see Warnings and Precautions (5.1) ] . In patients with cirrhosis Child-Pugh Class B and C, there is a potential increase in AQVESME exposure [see Clinical Pharmacology (12.3) ].
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
5.1 Hepatocellular Injury
AQVESME can cause hepatocellular injury. Avoid use of AQVESME in patients with cirrhosis. In patients with thalassemia treated with AQVESME, liver injury with and without jaundice has been observed within the first 6 months of exposure. Obtain liver tests (including ALT, AST, alkaline phosphatase, total bilirubin with fractionation) prior to the initiation of AQVESME, then every 4 weeks for the first 24 weeks, and as clinically indicated thereafter. Interrupt AQVESME if clinically significant increases in liver tests are observed or alanine aminotransferase is >5 times the upper limit of normal (ULN). Complete a comprehensive evaluation to rule out other causes of liver injury when drug-induced liver injury (DILI) is suspected. Discontinue AQVESME if hepatocellular injury due to AQVESME is suspected [see Dosage and Administration (2.3) ] .
Symptoms and signs of early liver injury may mimic those of thalassemia. Advise patients to report new or worsening symptoms of loss of appetite, nausea, right upper quadrant abdominal pain, vomiting, scleral icterus, jaundice, or dark urine while on AQVESME treatment.
During the double-blind period, 2 of 301 patients (0.66%) with thalassemia treated with AQVESME experienced adverse reactions suggestive of hepatocellular injury. Three additional patients experienced adverse reactions suggestive of hepatocellular injury during the open-label extension periods after switching from placebo to AQVESME. Of these 5 patients, two had serious liver injury and were hospitalized including 1 patient who developed jaundice (peak bilirubin 32 mg/dL). Another patient developed jaundice (peak bilirubin 4 mg/dL) without being hospitalized. These reactions were characterized by a time to onset within the first 6 months of treatment with peak elevations of alanine aminotransferase of >5×ULN with or without jaundice. All patients discontinued treatment with AQVESME, and these reactions improved upon treatment discontinuation.
AQVESME is available only through a restricted program under a REMS [see Warnings and Precautions (5.2) ] .
5.2 AQVESME REMS
AQVESME is available only through a restricted program under a REMS called the AQVESME REMS because of the risk of hepatocellular injury.
Notable requirements of the AQVESME REMS include the following:
- Prescribers must be certified by enrolling in the REMS and completing training.
- Prescribers must counsel patients receiving AQVESME about the risk of hepatocellular injury.
- Prescribers must monitor liver tests (including ALT, AST, alkaline phosphatase, total bilirubin with fractionation, and other tests as clinically indicated) to determine if the patient is appropriate to receive AQVESME treatment.
- Patients must enroll in the REMS and comply with the monitoring requirements.
- Pharmacies must be certified by enrolling in the REMS and must only dispense to patients who are authorized to receive AQVESME.
Further information is available at www.aqvesmerems.com or 1-800-625-9951.
ADVERSE REACTIONS
The following clinically significant adverse reaction is described elsewhere in labeling:
- Hepatocellular Injury [see Warnings and Precautions (5.1) ].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Alpha- and Beta-Thalassemia
A total of 301 patients with thalassemia received AQVESME, administered at 100 mg orally twice daily, for up to 59.9 weeks in the ENERGIZE trial (N=129) and the ENERGIZE-T trial (N=172) [see Clinical Studies (14) ] .
ENERGIZE Trial
Patients with non-transfusion-dependent thalassemia received AQVESME (N=129) or placebo (N=63).
The most common adverse reactions (≥5% and at least 5% higher in the AQVESME arm) in patients with non-transfusion-dependent thalassemia were headache and insomnia.
ENERGIZE-T Trial
Patients with transfusion-dependent thalassemia received AQVESME (N=172) or placebo (N=85).
The most common adverse reactions (≥5% and at least 5% higher in the AQVESME arm) in patients with transfusion-dependent thalassemia were headache and insomnia.
Serious adverse reactions occurred in 1.3% of patients with thalassemia treated with AQVESME, including supraventricular arrhythmia and supraventricular tachycardia. Permanent discontinuations of AQVESME due to an adverse reaction occurred in 1.3% of patients and included elevated hepatic transaminases and insomnia.
Table 1 summarizes the adverse reactions in the ENERGIZE and the ENERGIZE-T trials, individually and combined.
| ENERGIZE (Non-transfusion-dependent) | ENERGIZE-T (Transfusion-dependent) | Total | ||||
| Adverse Reactions | AQVESME (N=129) n (%) | Placebo (N=63) n (%) | AQVESME (N=172) n (%) | Placebo (N=85) n (%) | AQVESME (N=301) n (%) | Placebo (N=148) n (%) |
| Headache | 29 (22.5) | 6 (9.5) | 46 (26.7) | 10 (11.8) | 75 (24.9) | 16 (10.8) |
| Insomnia b | 35 (27.1) | 5 (7.9) | 38 (22.1) | 8 (9.4) | 73 (24.3) | 13 (8.8) |
| a Included adverse reactions that occurred in at least 5% of patients in the AQVESME arm and at least 5% higher than the placebo arm. b Term includes initial insomnia, middle insomnia, and terminal insomnia. | ||||||
Variations in Reproductive Hormones
Increases in serum testosterone (T) concentrations and decreases in serum estradiol (E2) concentrations were observed in men receiving AQVESME (Table 2). These changes in hormones were maintained during treatment with AQVESME. In 3 male patients who discontinued AQVESME and in whom reproductive hormone data were available following discontinuation of AQVESME, the hormone changes were reversible. In female patients, sex hormone analysis was limited due to physiologic variations in hormones during the menstrual cycle and the use of hormonal contraceptives.
| ENERGIZE (Non-transfusion-dependent) | ENERGIZE-T (Transfusion-dependent) | |||
| Parameter | AQVESME (46 males) | Placebo (25 males) | AQVESME (64 males) | Placebo (31 males) |
| Reproductive hormone analyses | ||||
| Testosterone (T) | ||||
Serum T concentration (mean) Baseline Change from baseline | 613 ng/dL 228 ng/dL | 505 ng/dL -2.8 ng/dL | 625 ng/dL 108 ng/dL | 666 ng/dL 66 ng/dL |
Serum T increased a Baseline Change from baseline | 2.2% 18.6% | 4.3% 0% | 3.3% 13.5% | 10% 6.9% |
| Estradiol (E2) | ||||
Serum E2 concentration (mean) Baseline Change from baseline | 29.6 pg/mL -8.7 pg/mL | 27.3 pg/mL 0 pg/mL | 26.4 pg/mL -5.0 pg/mL | 28.9 pg/mL 1.6 pg/mL |
| Serum E2 decreased b Baseline Change from baseline | 0% 2.5% | 9.5% 0% | 10.2% 8.5% | 6.9% 3.6% |
| a Percentage of subjects with serum T concentration above the upper limit of normal (greater than 1050 ng/dL) at baseline and percentage of subjects with serum T increases from baseline to above the upper limit of normal where baseline was within normal limits. b Percentage of subjects with serum E2 concentration below the lower limit of normal at baseline and percentage of subjects with serum E2 decreases from baseline to below the lower limit of normal where baseline was within normal limits. Note: Results from the ENERGIZE-T study do not include data from patients who received concomitant testosterone replacement therapies. | ||||
DRUG INTERACTIONS
7.1 Effect of Other Drugs on AQVESME
| Strong CYP3A Inhibitors | |
| Clinical Impact |
|
| Prevention or Management |
|
| Moderate CYP3A Inhibitors | |
| Clinical Impact |
|
| Prevention or Management |
|
| Strong CYP3A Inducers | |
| Clinical Impact |
|
| Prevention or Management |
|
| Moderate CYP3A Inducers | |
| Clinical Impact |
|
| Prevention or Management |
|
7.2 Effect of AQVESME on Other Drugs
| CYP3A Substrates | |
| Clinical Impact |
|
| Prevention or Management |
|
| CYP2B6 and CYP2C Substrates | |
| Clinical Impact |
|
| Prevention or Management |
|
| UGT1A1 Substrates | |
| Clinical Impact |
|
| Prevention or Management |
|
| P-gp Substrates | |
| Clinical Impact |
|
| Prevention or Management |
|
DESCRIPTION
The active ingredient of AQVESME is mitapivat, a pyruvate kinase activator, present as mitapivat sulfate. The chemical name of mitapivat sulfate is 8-quinolinesulfonamide, N-[4-[[4- (cyclopropylmethyl)-1-piperazinyl]carbonyl]phenyl]-, sulfate, hydrate (2:1:3). The chemical structure of mitapivat sulfate is:
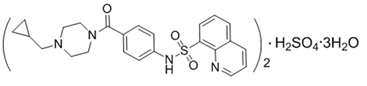
The molecular formula is (C24H26N4SO3)2 • H2SO4 • 3H2O, and the molecular weight is 1053.23 for mitapivat sulfate. Mitapivat sulfate is a white to off-white solid and is slightly soluble in water.
AQVESME is available as 100 mg tablets for oral administration. Each tablet contains 100 mg mitapivat free base, provided as 117.0 mg of the sulfate hydrate salt, and the following inactive ingredients: croscarmellose sodium, mannitol, microcrystalline cellulose, and sodium stearyl fumarate.
The 100 mg tablet film coating contains the inactive ingredients FD&C Blue No. 2, hypromellose, lactose monohydrate, titanium dioxide, triacetin, and macrogol/PEG. The tablets are imprinted with blue ink containing the inactive ingredients ammonium hydroxide, FD&C Blue No. 1, isopropyl alcohol, n-butyl alcohol, propylene glycol, shellac glaze, and titanium dioxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Mitapivat is a pyruvate kinase activator that acts by allosterically binding to the pyruvate kinase tetramer and increasing pyruvate kinase (PK) activity. Imbalances in globin chain production during erythropoiesis result in increased oxidative stress, which leads to ineffective erythropoiesis and hemolysis. In nonclinical models of beta-thalassemia, mitapivat improved energy homeostasis, RBC longevity, ineffective erythropoiesis, and hemolysis by increasing PK activity.
Pharmacodynamics
Mitapivat decreases 2,3 diphosphoglycerate (2,3-DPG) and increases ATP in healthy volunteers and in patients with thalassemia.
Cardiac Electrophysiology
At a dose 3 times the recommended dose, mitapivat did not prolong the QT interval to any clinically relevant extent.
Pharmacokinetics
The population pharmacokinetic model simulated C max , C trough , AUC 0-12 and accumulation ratio of mitapivat at the recommended dosage is listed in Table 3.
| Mitapivat Dosage | C max (ng/mL) | C trough (ng/mL) | AUC 0-12 (ng•h/mL) | Accumulation Ratio |
| 100 mg twice daily b | 1641.7 (12.9%) | 71 (18.5%) | 4835.6 (5.8%) | 0.83 |
| a Pharmacokinetic parameters are presented as geometric mean (CV%). The interval of the last 12 hours was selected for steady state PK parameters calculation. Residual error was not included during simulation. b The simulations were performed at steady state. | ||||
Absorption
Median t max values at steady state were 0.5 to 1.0 hour post-dose at 100 mg twice daily.
The absolute bioavailability after a single dose was approximately 73%.
Effect of Food
Following administration of a single dose of AQVESME in healthy subjects, a high-fat meal (approximately 900 to 1,000 total calories, with 500 to 600 calories from fat, 250 calories from carbohydrate, and 150 calories from protein) did not change the exposure (AUC inf ) of mitapivat, but reduced the rate of mitapivat absorption, with a 42% reduction in C max and a delay in t max of 2.3 hours when compared to dosing under fasted conditions.
Distribution
Mitapivat is highly protein bound (97.7%) in plasma with low RBC distribution (RBC-to-plasma ratio of 0.37). The mean volume of distribution at steady state (V ss ) was 42.5 L.
Elimination
Population pharmacokinetics derived median CL/F at steady state was 17.7 L/h at 100 mg twice daily.
Metabolism
In vitro studies showed that mitapivat is primarily metabolized by CYP3A4. Following a single oral dose of 120 mg of radiolabeled mitapivat to healthy subjects, unchanged mitapivat was the major circulating component.
Excretion
After a single oral administration of radiolabeled mitapivat to healthy subjects, the total recovery of administered radioactive dose was 89.2%, with 49.6% in the urine (2.6% unchanged) and 39.6% in the feces (<1% unchanged).
Specific Populations
No clinically meaningful effects on the pharmacokinetics of mitapivat were observed based on age, sex, race, or body weight.
Pediatric Population
The pharmacokinetics of mitapivat in children and adolescents (˂18 years old) have not been studied.
Hepatic Impairment
Mitapivat undergoes extensive hepatic metabolism. The pharmacokinetics of mitapivat were studied in adult subjects with moderate hepatic impairment (Child-Pugh Class B). After a single oral administration of 50 mg mitapivat, subjects with moderate hepatic impairment demonstrated 36% greater exposure (AUC ∞ ) to mitapivat, compared to subjects with normal hepatic function. Geometric mean C max values were similar between the groups. There were no major changes to plasma protein binding or elimination half-life in subjects with moderate hepatic impairment relative to healthy controls. The pharmacokinetics of mitapivat in subjects with severe hepatic impairment (Child-Pugh Class C) have not been studied.
Renal Impairment
The effects of renal impairment on mitapivat pharmacokinetics were assessed with population pharmacokinetic analyses. Steady state AUC of mitapivat in patients with eGFR 60 to <90 mL/min/1.73 m 2 was not significantly different compared to patients with eGFR ≥90 mL/min/1.73 m 2 . There are limited data available in patients with eGFR 30 to <60 mL/min/1.73 m 2 and no data available in patients with eGFR <30 mL/min/1.73 m 2 .
Drug Interaction Studies
Clinical Studies and Model-Based Approaches
Effect of Strong CYP3A Inhibitors on AQVESME
Itraconazole (a strong CYP3A inhibitor) increased mitapivat AUC inf and C max by 4.9-fold and 1.7-fold, respectively, following a single AQVESME dose of 20 mg. Itraconazole increased mitapivat AUC 0-12 and C max by 1.9‑fold and 1.6-fold, respectively, following AQVESME 100 mg twice daily. Ketoconazole (a strong CYP3A inhibitor) increased mitapivat AUC 0-12 and C max by approximately 3.9-fold and 2.4-fold, respectively, following AQVESME 100 mg twice daily.
Effect of Moderate CYP3A Inhibitors on AQVESME
Fluconazole (a moderate CYP3A inhibitor) increased mitapivat AUC 0-12 and C max by 2.7-fold and 1.7-fold, respectively, following AQVESME 100 mg twice daily.
Effect of Strong CYP3A Inducers on AQVESME
Rifampin (a strong CYP3A inducer) decreased mitapivat AUC inf and C max by 91% and 77%, respectively, following a single AQVESME dose of 50 mg. Rifampin decreased mitapivat AUC 0-12 and C max by 93% and 82%, respectively, following AQVESME 100 mg twice daily.
Effect of Moderate CYP3A Inducers on AQVESME
Efavirenz (a moderate CYP3A4 inducer) decreased mitapivat AUC 0-12 and C max by 52% and 21%, respectively, following AQVESME 100 mg twice daily.
Effect of AQVESME on CYP3A substrates
Midazolam (a CYP3A substrate) AUC inf and C max decreased by 65% and 59%, respectively, with AQVESME 100 mg twice daily.
Effect of AQVESME on P-gp Substrates
Co-administration of AQVESME with drugs that are substrates of P-gp may result in a clinically relevant increase in plasma concentrations of these substrates.
In vitro Studies
CYP450 and UGT Enzymes
Mitapivat induces CYP2B6, CYP2C8, CYP2C9, CYP2C19, and UGT1A1 .
Drug Transporter Systems
Mitapivat is a substrate and an inhibitor of P-gp.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Mitapivat was not carcinogenic in transgenic rasH2 mice up to the highest doses tested at 500 mg/kg/day in males and at 250 mg/kg/day in females when given orally for 26 weeks.
Mitapivat was not carcinogenic in rats when given orally up to 300 mg/kg/day in males and 200 mg/kg/day in females, at systemic exposures 35 times and >86 times the MRHD, respectively, based on AUC.
Mutagenesis
Mitapivat was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay. Mitapivat was not clastogenic in an in vitro human lymphocyte micronucleus assay or in an in vivo rat bone marrow micronucleus assay.
Fertility
In a fertility and early embryonic development study, oral administration of mitapivat twice daily in male rats prior to and during mating at doses up to 300 mg/kg/day, which represents 34 times the MRHD of 100 mg twice daily, based on AUC, did not result in adverse effects on fertility or reproductive function. In female rats, twice daily oral administration of mitapivat prior to mating and continuing through organogenesis, at doses up to 200 mg/kg/day, which represents 37 times the MRHD of 100 mg twice daily, based on AUC, did not result in adverse effects on fertility or reproductive function.
CLINICAL STUDIES
Patients with Transfusion-Dependent and Non-Transfusion-Dependent alpha- or beta-Thalassemia
Transfusion-Dependent alpha- or beta-Thalassemia
The efficacy of AQVESME was evaluated in ENERGIZE-T, a multinational, randomized, double-blind, placebo-controlled clinical study (NCT04770779) of 258 adult patients with transfusion-dependent alpha- or beta-thalassemia, defined as having had 6 to 20 RBC units transfused and no longer than a 6-week transfusion-free period during the 24 weeks prior to randomization. Patients were included if they had a documented diagnosis of thalassemia (beta-thalassemia with or without alpha-globin gene mutations, HbE/beta-thalassemia, or alpha-thalassemia/HbH disease). Randomization was stratified by geographical region (North America and Europe vs Asia-Pacific vs Rest of World) and thalassemia genotype (β 0 /β 0 vs non-β 0 /β 0 ).
Among the 258 patients with transfusion-dependent alpha- or beta-thalassemia, 171 patients were randomized to receive 100 mg of AQVESME twice daily during the 48-week double-blind period.
The median duration of treatment with AQVESME was 48.1 weeks (range: 0.3 to 59.9 weeks). Overall, 104 (60.5%) patients were exposed to AQVESME for >48 weeks. Among the 258 randomized patients, the median age was 33.5 years (range: 18 to 67) and 47.3% were male; 62.0% were from North America and Europe, 18.2% were from Asia-Pacific, and 19.8% were from the rest of the world; race was reported in 95.7% of patients: 60.1% White, 30.2% Asian, 0.8% Black or African American, 0.8% Multiracial, and 3.9% unknown.
The baseline disease characteristics are shown in Table 4.
| Baseline Disease Characteristics | AQVESME N=171 | Placebo N=87 | Total N=258 |
| Hemoglobin (g/dL) , n a Median (min, max) | 9.0 (5.1, 11.8) | 8.9 (5.1, 10.9) | 8.9 (5.1, 11.8) |
| Thalassemia Genotype , n (%) beta 0 / beta 0 non-beta 0 / beta 0 | 75 (44) 96 (56) | 39 (45) 48 (55) | 114 (44) 144 (56) |
| Transfusion Burden (RBC units) , n (%) b ≤12 >12 | 54 (32) 117 (68) | 21 (24) 66 (76) | 75 (29) 183 (71) |
| Hepatic Iron Concentration (mg/g) , n Median (min, max) | 133 4.58 (0.37, 28.21) | 74 4.43 (0.37, 20.47) | 207 4.55 (0.37, 28.21) |
| Prior History of Splenectomy, n (%) | 92 (54) | 49 (56) | 141 (55) |
| Prior History of Cholecystectomy, n (%) | 42 (25) | 24 (28) | 66 (26) |
| Prior History of Iron Chelation, n (%) | 165 (96) | 87 (100) | 252 (98) |
| Prior History of Hydroxyurea, n (%) | 7 (4.1) | 3 (3.4) | 10 (3.9) |
| Hb: hemoglobin, RBC: red blood cells a Pretransfusion Hb threshold is the mean of all pretransfusion Hb concentrations for the RBC transfusions administered during the 24-week period before randomization. b Total number of RBC units transfused in the 24-week period before randomization. | |||
Efficacy was based upon transfusion reduction response, defined as ≥50% reduction in the number of red blood cell units transfused with a reduction of at least 2 units of RBCs transfused in any consecutive 12-week period through Week 48 compared with baseline. The efficacy results are shown in Table 5.
| AQVESME N= 171 | Placebo N=87 | Difference | ||
| Endpoints | n (%) | n (%) | Adjusted Rate Difference a (%) (95% CI) | p-value b |
| ≥50% reduction from baseline in RBC units transfused in any consecutive 12 weeks, with a reduction of at least 2 units | 52 (30.4) | 11 (12.6) | 17.6 (8.0, 27.2) | 0.0003 |
| Endpoints | n (%) | n (%) | Adjusted Rate Difference b (%) (95% CI) | p-value a |
| ≥50% reduction from baseline in RBC units transfused in any consecutive 24 weeks | 23 (13.5) | 2 (2.3) | 11.1 (5.1, 17.0) | 0.0003 |
| ≥33% reduction from baseline in RBC units from Week 13 through Week 48 | 25 (14.6) | 1 (1.1) | 13.4 (7.7, 19.1) | <0.0001 |
| ≥50% reduction from baseline in RBC units from Week 13 through Week 48 | 13 (7.6) | 1 (1.1) | 6.4 (1.9, 10.9) | 0.0056 |
| CI: confidence interval, RBC: red blood cell a All p-values are 2-sided and all results are statistically significant. b The difference is adjusted for randomization stratification factors, which included geographical region (North America and Europe vs Asia-Pacific vs Rest of World) and thalassemia genotype (β 0 /β 0 vs non-β 0 /β 0 ). The two-sided p-value is based on the Mantel-Haenszel stratum weighted method adjusting for the randomization stratification factors. | ||||
Non-Transfusion-Dependent alpha- or beta-Thalassemia
The efficacy of AQVESME was evaluated in ENERGIZE, a multinational, randomized, double-blind, placebo-controlled clinical study (NCT04770753) of 194 adults with non-transfusion-dependent alpha- or beta-thalassemia, defined as having had no more than 5 RBC units transfused during the 24‑week period prior to randomization and no RBC transfusions within 8 weeks prior to informed consent and during the screening period. Patients were included if they had a documented diagnosis of thalassemia (beta-thalassemia with or without alpha-globin gene mutations, HbE/beta-thalassemia, or alpha-thalassemia/HbH disease) and a baseline Hb concentration ≤10 g/dL. Randomization was stratified by baseline Hb concentrations (≤9 g/dL vs 9.1-10 g/dL) and thalassemia genotype (alpha-thalassemia/HbH disease vs beta-thalassemia).
Among the 194 patients with non-transfusion-dependent alpha- or beta-thalassemia, 130 patients were randomized to receive 100 mg of AQVESME twice daily during the 24-week double-blind period.
The median duration of treatment with AQVESME was 24.1 weeks (range: 1.1 to 28.1 weeks). Overall, 97 (75%) patients were exposed to AQVESME for >24 weeks. Among the 194 randomized patients, the median age was 41 years (range: 18 to 69) and 36.6% were male; race was reported in 99% of patients: 56.2% White, 39.2% Asian, 1.0% Black or African American, 0.5% Multiracial, and 2.1% unknown.
The baseline disease characteristics are shown in Table 6.
| Baseline Disease Characteristics a | AQVESME N=130 | Placebo N=64 | Total N=194 |
| Hemoglobin (g/dL) , n Median (min, max) | 130 8.4 (5.3, 10.4) | 64 8.4 (5.9, 10.7) | 194 8.4 (5.3, 10.7) |
| Thalassemia Genotype , n (%) alpha-thalassemia/HbH disease beta-thalassemia | 42 (32) 88 (68) | 20 (31) 44 (69) | 62 (32) 132 (68) |
| Transfusion Burden (RBC units) b , n (%) 0 1-2 3-5 | 114 (87.7) 10 (7.7) 6 (4.6) | 54 (84.4) 7 (10.9) 3 (4.7) | 168 (86.6) 17 (8.8) 9 (4.6) |
| Reticulocyte (Fraction of 1) , n Median (min, max) | 122 0.05 (0.00, 0.30) | 58 0.04 (0.00, 0.22) | 180 0.05 (0.00, 0.30) |
| Indirect Bilirubin (mg/dL) , n Median (min, max) | 130 1.37 (0.13, 9.11) | 62 1.32 (0.15, 4.77) | 192 1.34 (0.13, 19.11) |
| LDH (U/L) , n Median (min, max) | 130 264 (108, 1208) | 64 267 (110, 1009) | 194 265 (108, 1208) |
| Hepatic Iron Concentration (mg/g) , n Median (min, max) | 98 3.93 (0.75, 27.19) | 52 2.76 (0.75, 18.53) | 150 3.64 (0.75, 27.19) |
| Prior History of Splenectomy, n (%) | 47 (36) | 25 (39) | 72 (37) |
| Prior History of Cholecystectomy, n (%) | 45 (35) | 16 (25) | 61 (31) |
| Prior History of Iron Chelation, n (%) | 46 (35) | 22 (34) | 68 (35) |
| Prior History of Hydroxyurea, n (%) | 11 (8.5) | 6 (9.4) | 17 (8.8) |
| Hb: hemoglobin, LDH: lactate dehydrogenase, RBC: red blood cell a n is the number of patients with non-missing data. b Total number of RBC units transfused in the 24-week period before randomization. | |||
Efficacy was based upon Hb response, defined as a ≥1 g/dL increase in average Hb concentration from Week 12 through Week 24 compared with baseline and a mean change from baseline in fatigue-related symptoms and impacts assessed by a patient-reported outcome instrument, the Functional Assessment of Chronic Illness Therapy – Fatigue Scale (FACIT-Fatigue). See Table 7 for efficacy results.
| AQVESME N= 130 | Placebo N=64 | Difference | ||
| Endpoint | n (%) | n (%) | Adjusted Rate Difference (%) (95% CI) | p-value a |
| Hb Response b | 55 (42.3) | 1 (1.6) | 40.9 (32.0, 49.8) | <0.0001 |
| Endpoint | LS mean (95% CI) | LS mean (95% CI) | LS mean difference (95% CI) | p-value a |
| Hemoglobin (g/dL) c | 0.86 (0.73, 0.99) | -0.11 (-0.28, 0.07) | 0.96 (0.78, 1.15) | <0.0001 |
| FACIT-Fatigue d | 4.85 (3.41, 6.30) | 1.46 (-0.43, 3.34) | 3.40 (1.21, 5.59) | 0.0026 |
| CI: confidence interval, Hb: hemoglobin, LS: least squares a All p-values are 2-sided and all results are statistically significant. b For Hb response, the difference is adjusted for randomization stratification factors, which included Hb concentrations (≤9.0 g/dL vs 9.1-10.0 g/dL) and thalassemia genotype (alpha-thalassemia/HbH disease vs beta-thalassemia). The two‑sided p-value is based on the Mantel-Haenszel stratum weighted method adjusting for the randomization stratification factors. c Change from baseline in average Hb concentration from Week 12 through Week 24. d Change from baseline in average FACIT–Fatigue score from Week 12 through Week 24. FACIT-Fatigue total scores range from 0 to 52 with higher scores indicating less fatigue. At baseline, patients reported a mean FACIT-Fatigue score of approximately 36 in both the AQVESME and placebo arms. Note: For the endpoints of change from baseline in average Hb concentration from Week 12 through Week 24 and change from baseline in average FACIT-Fatigue score from Week 12 through Week 24, the 95% CIs and the two-sided p-values are based on an analysis of covariance (ANCOVA) model, which included change from baseline as the dependent variable, treatment group as the independent variable, and baseline and the randomization stratification factors as covariates. The results are based on observed cases. | ||||
Eighty-seven percent of patients in the AQVESME arm experienced an increase from baseline in average Hb from Weeks 12 through 24 (see Figure 1). Figure 2 depicts change from baseline in hemoglobin over time.
Figure 1: Average Change from Baseline in Hemoglobin from Week 12 through Week 24 by Patient – All Randomized Patients (ENERGIZE)
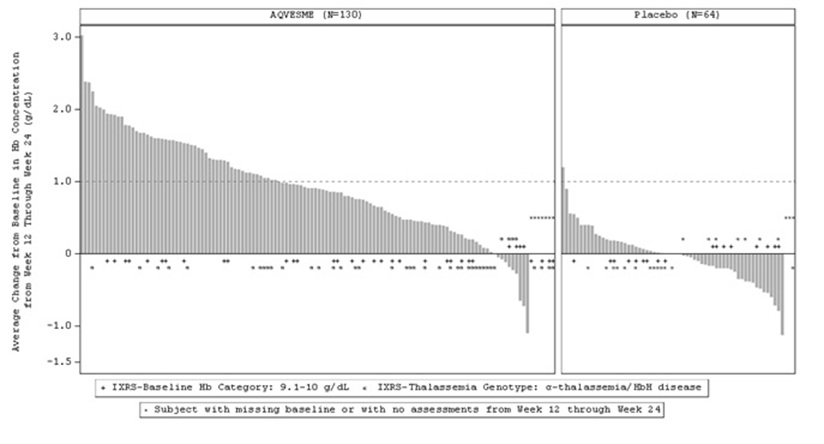
Figure 2: Change from Baseline in Hemoglobin Over Time – All Randomized Patients (ENERGIZE)
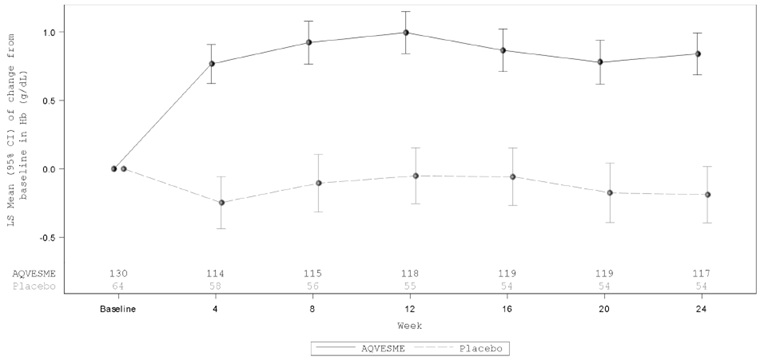
Of the 55 patients with Hb response in the AQVESME arm, the average increase in Hb was 1.6 g/dL and the median duration of response was 19.6 weeks (range: 4.0 to 23.4+ weeks) during the 24-week double-blind period.
Patients in the AQVESME arm experienced an improvement compared to placebo in the change from baseline to Week 24 for 2 markers of hemolysis (indirect bilirubin [−0.62 mg/dL (95% CI: −0.80, −0.44)] and lactate dehydrogenase [−24.28 U/L (95% CI: −45.40, −3.15)]).
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
| AQVESME 28-Day Packs | ||||
| Tablet Strength | Package Configuration | Tablet Description | Tablet Imprint | NDC |
| 100 mg | Carton containing 4 blister wallets. Each blister wallet contains 14 tablets. | Oblong, blue, film-coated tablets | "M100" printed on both sides | Carton: 71334-235-00 Blister wallet: 71334-235-14 |
Storage
Store in a refrigerator 2°C to 8°C (36°F to 46°F).
AQVESME may be stored at room temperature, 20°C to 25°C (68°F to 77°F) for up to 3 months. The discard date is 3 months after removal of the tablets from the refrigerator. Write the discard date in the space provided on the blister wallet and/or the carton. Discard the tablets if not used within 3 months or if the expiration date has passed, whichever occurs first. Dispense and store the blister wallets in the original carton until use.
Mechanism of Action
Mitapivat is a pyruvate kinase activator that acts by allosterically binding to the pyruvate kinase tetramer and increasing pyruvate kinase (PK) activity. Imbalances in globin chain production during erythropoiesis result in increased oxidative stress, which leads to ineffective erythropoiesis and hemolysis. In nonclinical models of beta-thalassemia, mitapivat improved energy homeostasis, RBC longevity, ineffective erythropoiesis, and hemolysis by increasing PK activity.