Get your patient on Abacavir - Abacavir Sulfate solution (Abacavir Sulfate)
Abacavir - Abacavir Sulfate solution prescribing information
WARNING: HYPERSENSITIVITY REACTIONS
Serious and sometimes fatal hypersensitivity reactions, with multiple organ involvement, have occurred with abacavir.
Patients who carry the HLA-B•5701 allele are at a higher risk of a hypersensitivity reaction to abacavir; although, hypersensitivity reactions have occurred in patients who do not carry the HLA-B•5701 allele [see Warnings and Precautions (5.1) ] .
Abacavir is contraindicated in patients with a prior hypersensitivity reaction to abacavir and in HLA-B•5701-positive patients [see Contraindications (4) , Warnings and Precautions (5.1) ] . All patients should be screened for the HLA-B•5701 allele prior to initiating therapy with abacavir or reinitiation of therapy with abacavir, unless patients have a previously documented HLA-B•5701 allele assessment. Discontinue abacavir immediately if a hypersensitivity reaction is suspected, regardless of HLA-B•5701 status and even when other diagnoses are possible [see Contraindications (4) , Warnings and Precautions (5.1) ].
Following a hypersensitivity reaction to abacavir, NEVER restart abacavir or any other abacavir-containing product because more severe symptoms, including death can occur within hours. Similar severe reactions have also occurred rarely following the reintroduction of abacavir-containing products in patients who have no history of abacavir hypersensitivity [see Warnings and Precautions (5.1) ] .
INDICATIONS AND USAGE
Abacavir oral solution, in combination with other antiretroviral agents, is indicated for the treatment of human immunodeficiency virus (HIV-1) infection.
DOSAGE AND ADMINISTRATION
- Before initiating abacavir oral solution, screen for the HLA-B•5701 allele. (2.1 )
- Adults: 600 mg daily, administered as either 300 mg twice daily or 600 mg once daily. (2.2 )
- Pediatric Patients Aged 3 Months and Older: Administered either once or twice daily. Dose should be calculated on body weight (kg) and should not exceed 600 mg daily. (2.3 )
- Patients with Hepatic Impairment: Mild hepatic impairment – 200 mg twice daily. (2.4)
Screening for HLA-B•5701 Allele prior to Starting Abacavir Oral Solution
Screen for the HLA-B•5701 allele prior to initiating therapy with abacavir oral solution [see Boxed Warning , Warnings and Precautions (5.1) ] .
Recommended Dosage for Adult Patients
The recommended dosage of abacavir oral solution for adults is 600 mg daily, administered orally as either 300 mg twice daily or 600 mg once daily, in combination with other antiretroviral agents.
Recommended Dosage for Pediatric Patients
The recommended dosage of abacavir oral solution in HIV-1-infected pediatric patients aged 3 months and older is 8 mg per kg orally twice daily or 16 mg per kg orally once daily (up to a maximum of 600 mg daily) in combination with other antiretroviral agents.
Recommended Dosage for Patients with Hepatic Impairment
The recommended dose of abacavir oral solution in patients with mild hepatic impairment (Child-Pugh Class A) is 200 mg twice daily. To enable dose reduction, abacavir oral solution (10 mL twice daily) should be used for the treatment of these patients. The safety, efficacy, and pharmacokinetic properties of abacavir have not been established in patients with moderate to severe hepatic impairment; therefore, abacavir oral solution is contraindicated in these patients.
DOSAGE FORMS AND STRENGTHS
Abacavir oral solution USP, each mL containing abacavir sulfate USP equivalent to 20 mg of abacavir, is a clear to opalescent, yellowish, strawberry-banana-flavored liquid.
USE IN SPECIFIC POPULATIONS
- Lactation: Women infected with HIV should be instructed not to breastfeed due to potential for HIV transmission. (8.2 )
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to abacavir during pregnancy. Healthcare Providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Available data from the APR show no difference in the overall risk of birth defects for abacavir compared with the background rate for birth defects of 2.7% in the Metropolitan Atlanta Congenital Defects Program (MACDP) reference population (see Data). The APR uses the MACDP as the U.S. reference population for birth defects in the general population. The MACDP evaluates women and infants from a limited geographic area and does not include outcomes for births that occurred at less than 20 weeks' gestation. The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15% to 20%. The background risk for major birth defects and miscarriage for the indicated population is unknown.
In animal reproduction studies, oral administration of abacavir to pregnant rats during organogenesis resulted in fetal malformations and other embryonic and fetal toxicities at exposures 35 times the human exposure (AUC) at the recommended clinical daily dose. However, no adverse developmental effects were observed following oral administration of abacavir to pregnant rabbits during organogenesis, at exposures approximately 9 times the human exposure (AUC) at the recommended clinical dose (see Data).
Data
Human Data: Based on prospective reports to the APR of exposures to abacavir during pregnancy resulting in live births (including over 1,300 exposed in the first trimester and over 1,300 exposed in second/third trimester), there was no difference between the overall risk of birth defects for abacavir compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of defects in live births was 3.2% (95% CI: 2.3% to 4.3%) following first trimester exposure to abacavir-containing regimens and 2.9% (95% CI: 2.1% to 4.0%) following second/third trimester exposure to abacavir-containing regimens. Abacavir has been shown to cross the placenta and concentrations in neonatal plasma at birth were essentially equal to those in maternal plasma at delivery [see Clinical Pharmacology (12.3) ].
Animal Data: Abacavir was administered orally to pregnant rats (at 100, 300, and 1,000 mg per kg per day) and rabbits (at 125, 350, or 700 mg per kg per day) during organogenesis (on Gestation Days 6 through 17 and 6 through 20, respectively). Fetal malformations (increased incidences of fetal anasarca and skeletal malformations) or developmental toxicity (decreased fetal body weight and crown-rump length) were observed in rats at doses up to 1,000 mg per kg per day, resulting in exposures approximately 35 times the human exposure (AUC) at the recommended daily dose. No developmental effects were observed in rats at 100 mg per kg per day, resulting in exposures (AUC) 3.5 times the human exposure at the recommended daily dose. In a fertility and early embryo-fetal development study conducted in rats (at 60, 160, or 500 mg per kg per day), embryonic and fetal toxicities (increased resorptions, decreased fetal body weights) or toxicities to the offspring (increased incidence of stillbirth and lower body weights) occurred at doses up to 500 mg per kg per day. No developmental effects were observed in rats at 60 mg per kg per day, resulting in exposures (AUC) approximately 4 times the human exposure at the recommended daily dose. Studies in pregnant rats showed that abacavir is transferred to the fetus through the placenta. In pregnant rabbits, no developmental toxicities and no increases in fetal malformations occurred at up to the highest dose evaluated, resulting in exposures (AUC) approximately 9 times the human exposure at the recommended dose.
Lactation
Risk Summary
The Centers for Disease Control and Prevention recommends that HIV-1-infected mothers in the United States not breastfeed their infants to avoid risking postnatal transmission of HIV-1 infection. Abacavir is present in human milk. There is no information on the effects of abacavir on the breastfed infant or the effects of the drug on milk production. Because of the potential for (1) HIV-1 transmission (in HIV-negative infants), (2) developing viral resistance (in HIV-positive infants), and (3) adverse reactions in a breastfed infant, instruct mothers not to breastfeed if they are receiving abacavir.
Pediatric Use
The safety and effectiveness of abacavir have been established in pediatric patients aged 3 months and older. Use of abacavir is supported by pharmacokinetic trials and evidence from adequate and well-controlled trials of abacavir in adults and pediatric subjects [see Dosage and Administration (2.3) , Adverse Reactions (6.2) , Clinical Pharmacology (12.3) , Clinical Studies (14.2) ] .
Geriatric Use
Clinical trials of abacavir did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, caution should be exercised in the administration of abacavir in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Patients with Impaired Hepatic Function
A dose reduction is required for patients with mild hepatic impairment (Child-Pugh Class A) [see Dosage and Administration (2.4) ] . The safety, efficacy, and pharmacokinetic properties of abacavir have not been established in patients with moderate or severe hepatic impairment; therefore, abacavir is contraindicated in these patients [see Contraindications (4) , Clinical Pharmacology (12.3) ] .
CONTRAINDICATIONS
Abacavir oral solution is contraindicated in patients:
- who have the HLA-B•5701 allele [see Warnings and Precautions (5.1) ] .
- with prior hypersensitivity reaction to abacavir [see Warnings and Precautions (5.1) ].
- with moderate or severe hepatic impairment [see Use in Specific Populations (8.6) ] .
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions
Serious and sometimes fatal hypersensitivity reactions have occurred with abacavir. These hypersensitivity reactions have included multi-organ failure and anaphylaxis and typically occurred within the first 6 weeks of treatment with abacavir (median time to onset was 9 days); although abacavir hypersensitivity reactions have occurred any time during treatment [see Adverse Reactions (6.1) ] . Patients who carry the HLA-B•5701 allele are at a higher risk of abacavir hypersensitivity reactions; although, patients who do not carry the HLA-B•5701 allele have developed hypersensitivity reactions. Hypersensitivity to abacavir was reported in approximately 206 (8%) of 2,670 patients in 9 clinical trials with abacavir-containing products where HLA-B•5701 screening was not performed. The incidence of suspected abacavir hypersensitivity reactions in clinical trials was 1% when subjects carrying the HLA-B•5701 allele were excluded. In any patient treated with abacavir, the clinical diagnosis of hypersensitivity reaction must remain the basis of clinical decision making.
Due to the potential for severe, serious, and possibly fatal hypersensitivity reactions with abacavir:
- All patients should be screened for the HLA-B•5701 allele prior to initiating therapy with abacavir or reinitiation of therapy with abacavir, unless patients have a previously documented HLA-B•5701 allele assessment.
- Abacavir is contraindicated in patients with a prior hypersensitivity reaction to abacavir and in HLA-B•5701-positive patients.
- Before starting abacavir, review medical history for prior exposure to any abacavir- containing product. NEVER restart abacavir or any other abacavir-containing product following a hypersensitivity reaction to abacavir, regardless of HLA-B•5701 status.
- To reduce the risk of a life-threatening hypersensitivity reaction, regardless of HLA-B•5701 status, discontinue abacavir immediately if a hypersensitivity reaction is suspected, even when other diagnoses are possible (e.g., acute onset respiratory diseases such as pneumonia, bronchitis, pharyngitis, or influenza; gastroenteritis; or reactions to other medications).
- If a hypersensitivity reaction cannot be ruled out, do not restart abacavir or any other abacavir-containing products because more severe symptoms which may include life-threatening hypotension and death, can occur within hours.
- If a hypersensitivity reaction is ruled out, patients may restart abacavir. Rarely, patients who have stopped abacavir for reasons other than symptoms of hypersensitivity have also experienced life-threatening reactions within hours of reinitiating abacavir therapy. Therefore, reintroduction of abacavir or any other abacavir-containing product is recommended only if medical care can be readily accessed.
- A Medication Guide and Warning Card that provide information about recognition of hypersensitivity reactions should be dispensed with each new prescription and refill.
Lactic Acidosis and Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including abacavir. A majority of these cases have been in women. Female sex and obesity may be risk factors for the development of lactic acidosis and severe hepatomegaly with steatosis in patients treated with antiretroviral nucleoside analogues. Treatment with abacavir should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity, which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations.
Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including abacavir. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment. Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable and can occur many months after initiation of treatment.
Myocardial Infarction
Several prospective, observational, epidemiological studies have reported an association with the use of abacavir and the risk of myocardial infarction (MI). Meta-analyses of randomized, controlled, clinical trials have observed no excess risk of MI in abacavir-treated subjects as compared with control subjects. To date, there is no established biological mechanism to explain a potential increase in risk. In totality, the available data from the observational studies and from controlled clinical trials show inconsistency; therefore, evidence for a causal relationship between abacavir treatment and the risk of MI is inconclusive.
As a precaution, the underlying risk of coronary heart disease should be considered when prescribing antiretroviral therapies, including abacavir, and action taken to minimize all modifiable risk factors (e.g., hypertension, hyperlipidemia, diabetes mellitus, smoking).
ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Serious and sometimes fatal hypersensitivity reactions [see Boxed Warning , Warnings and Precautions (5.1) ] .
- Lactic acidosis and severe hepatomegaly with steatosis [see Warnings and Precautions (5.2) ] .
- Immune reconstitution syndrome [see Warnings and Precautions (5.3) ] .
- Myocardial infarction [see Warnings and Precautions (5.4) ] .
Clinical Trials Experience in Adult Subjects
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Serious and Fatal Abacavir-Associated Hypersensitivity Reactions
In clinical trials, serious and sometimes fatal hypersensitivity reactions have occurred with abacavir [see Boxed Warning , Warnings and Precautions (5.1) ]. These reactions have been characterized by 2 or more of the following signs or symptoms: (1) fever; (2) rash; (3) gastrointestinal symptoms (including nausea, vomiting, diarrhea, or abdominal pain); (4) constitutional symptoms (including generalized malaise, fatigue, or achiness); (5) respiratory symptoms (including dyspnea, cough, or pharyngitis). Almost all abacavir hypersensitivity reactions include fever and/or rash as part of the syndrome.
Other signs and symptoms have included lethargy, headache, myalgia, edema, arthralgia, and paresthesia. Anaphylaxis, liver failure, renal failure, hypotension, adult respiratory distress syndrome, respiratory failure, myolysis, and death have occurred in association with these hypersensitivity reactions. Physical findings have included lymphadenopathy, mucous membrane lesions (conjunctivitis and mouth ulcerations), and maculopapular or urticarial rash (although some patients had other types of rashes and others did not have a rash). There were reports of erythema multiforme. Laboratory abnormalities included elevated liver chemistries, elevated creatine phosphokinase, elevated creatinine, and lymphopenia, and abnormal chest x-ray findings (predominantly infiltrates, which were localized).
Additional Adverse Reactions with Use of Abacavir
Therapy-Naive Adults: Treatment-emergent clinical adverse reactions (rated by the investigator as moderate or severe) with a greater than or equal to 5% frequency during therapy with abacavir 300 mg twice daily, lamivudine 150 mg twice daily, and efavirenz 600 mg daily compared with zidovudine 300 mg twice daily, lamivudine 150 mg twice daily, and efavirenz 600 mg daily from CNA30024 are listed in Table 2.
| Adverse Reaction | Abacavir plus Lamivudine plus Efavirenz (n = 324) | Zidovudine plus Lamivudine plus Efavirenz (n = 325) |
| Dreams/sleep disorders Drug hypersensitivity Headaches/migraine Nausea Fatigue/malaise Diarrhea Rashes Abdominal pain/gastritis/gastrointestinal signs and symptoms Depressive disorders Dizziness Musculoskeletal pain Bronchitis Vomiting | 10% 9% 7% 7% 7% 7% 6% 6% 6% 6% 6% 4% 2% | 10% <1% b 11% 11% 10% 6% 12% 8% 6% 6% 5% 5% 9% |
a This trial used double-blind ascertainment of suspected hypersensitivity reactions. During the blinded portion of the trial, suspected hypersensitivity to abacavir was reported by investigators in 9% of 324 subjects in the abacavir group and 3% of 325 subjects in the zidovudine group.
b Ten (3%) cases of suspected drug hypersensitivity were reclassified as not being due to abacavir following unblinding.
Treatment-emergent clinical adverse reactions (rated by the investigator as moderate or severe) with a greater than or equal to 5% frequency during therapy with abacavir 300 mg twice daily, lamivudine 150 mg twice daily, and zidovudine 300 mg twice daily compared with indinavir 800 mg 3 times daily, lamivudine 150 mg twice daily, and zidovudine 300 mg twice daily from CNA3005 are listed in Table 3.
| Adverse Reaction | Abacavir plus Lamivudine/Zidovudine (n = 262) | Indinavir plus Lamivudine/Zidovudine (n = 264) |
| Nausea Headache Malaise and fatigue Nausea and vomiting Hypersensitivity reaction Diarrhea Fever and/or chills Depressive disorders Musculoskeletal pain Skin rashes Ear/nose/throat infections Viral respiratory infections Anxiety Renal signs/symptoms Pain (non-site-specific) | 19% 13% 12% 10% 8% 7% 6% 6% 5% 5% 5% 5% 5% <1% <1% | 17% 9% 12% 10% 2% 5% 3% 4% 7% 4% 4% 5% 3% 5% 5% |
Five subjects receiving abacavir in CNA3005 experienced worsening of pre-existing depression compared with none in the indinavir arm. The background rates of pre-existing depression were similar in the 2 treatment arms.
Abacavir Once Daily versus Abacavir Twice Daily (CNA30021): Treatment-emergent clinical adverse reactions (rated by the investigator as at least moderate) with a greater than or equal to 5% frequency during therapy with abacavir 600 mg once daily or abacavir 300 mg twice daily, both in combination with lamivudine 300 mg once daily and efavirenz 600 mg once daily from CNA30021, were similar. For hypersensitivity reactions, subjects receiving abacavir once daily showed a rate of 9% in comparison with a rate of 7% for subjects receiving abacavir twice daily. However, subjects receiving abacavir 600 mg once daily experienced a significantly higher incidence of severe drug hypersensitivity reactions and severe diarrhea compared with subjects who received abacavir 300 mg twice daily. Five percent (5%) of subjects receiving abacavir 600 mg once daily had severe drug hypersensitivity reactions compared with 2% of subjects receiving abacavir 300 mg twice daily. Two percent (2%) of subjects receiving abacavir 600 mg once daily had severe diarrhea while none of the subjects receiving abacavir 300 mg twice daily had this event.
Laboratory Abnormalities: Laboratory abnormalities (Grades 3 to 4) in therapy-naive adults during therapy with abacavir 300 mg twice daily, lamivudine 150 mg twice daily, and efavirenz 600 mg daily compared with zidovudine 300 mg twice daily, lamivudine 150 mg twice daily, and efavirenz 600 mg daily from CNA30024 are listed in Table 4.
| Grade 3/4 Laboratory Abnormalities | Abacavir plus Lamivudine plus Efavirenz (n = 324) | Zidovudine plus Lamivudine plus Efavirenz (n = 325) |
| Elevated CPK (>4 X ULN) Elevated ALT (>5 X ULN) Elevated AST (>5 X ULN) Hypertriglyceridemia (>750 mg/dL) Hyperamylasemia (>2 X ULN) Neutropenia (ANC <750/mm 3 ) Anemia (Hgb ≤6.9 gm/dL) Thrombocytopenia (Platelets <50,000/mm 3 ) Leukopenia (WBC ≤1,500/mm 3 ) | 8% 6% 6% 6% 4% 2% <1% 1% <1% | 8% 6% 5% 5% 5% 4% 2% <1% 2% |
ULN = Upper limit of normal.
n = Number of subjects assessed.
Laboratory abnormalities in CNA3005 are listed in Table 5.
| Grade 3/4 Laboratory Abnormalities | Abacavir plus Lamivudine/Zidovudine (n = 262) | Indinavir plus Lamivudine/Zidovudine (n = 264) |
| Elevated CPK (>4 x ULN) ALT (>5 x ULN) Neutropenia (<750/mm 3 ) Hypertriglyceridemia (>750 mg/dL) Hyperamylasemia (>2 x ULN) Hyperglycemia (>13.9 mmol/L) Anemia (Hgb ≤6.9 g/dL) | 18 (7%) 16 (6%) 13 (5%) 5 (2%) 5 (2%) 2 (<1%) 0 (0%) | 18 (7%) 16 (6%) 13 (5%) 3 (1%) 1 (<1%) 2 (<1%) 3 (1%) |
ULN = Upper limit of normal.
n = Number of subjects assessed.
The frequencies of treatment-emergent laboratory abnormalities were comparable between treatment groups in CNA30021.
Clinical Trials Experience in Pediatric Subjects
Therapy-Experienced Pediatric Subjects (Twice-Daily Dosing) Treatment-emergent clinical adverse reactions (rated by the investigator as moderate or severe) with a greater than or equal to 5% frequency during therapy with abacavir 8 mg per kg twice daily, lamivudine 4 mg per kg twice daily, and zidovudine 180 mg per m 2 twice daily compared with lamivudine 4 mg per kg twice daily and zidovudine 180 mg per m 2 twice daily from CNA3006 are listed in Table 6.
| Adverse Reaction | Abacavir plus Lamivudine plus Zidovudine (n = 102) | Lamivudine plus Zidovudine (n = 103) |
| Fever and/or chills Nausea and vomiting Skin rashes Ear/nose/throat infections Pneumonia Headache | 9% 9% 7% 5% 4% 1% | 7% 2% 1% 1% 5% 5% |
Laboratory Abnormalities: In CNA3006, laboratory abnormalities (anemia, neutropenia, liver function test abnormalities, and CPK elevations) were observed with similar frequencies as in a trial of therapy-naive adults (CNA30024). Mild elevations of blood glucose were more frequent in pediatric subjects receiving abacavir (CNA3006) as compared with adult subjects (CNA30024). Other Adverse Events In addition to adverse reactions and laboratory abnormalities reported in Tables 2, 3, 4, 5, and 6, other adverse reactions observed in the expanded access program were pancreatitis and increased GGT. Pediatric Subjects Once-Daily versus Twice-Daily Dosing (COL105677): The safety of once-daily compared with twice-daily dosing of abacavir was assessed in the ARROW trial. Primary safety assessment in the ARROW trial was based on Grade 3 and Grade 4 adverse events. The frequency of Grade 3 and 4 adverse events was similar among subjects randomized to once-daily dosing compared with subjects randomized to twice-daily dosing. One event of Grade 4 hepatitis in the once-daily cohort was considered as uncertain causality by the investigator and all other Grade 3 or 4 adverse events were considered not related by the investigator.
Postmarketing Experience
The following adverse reactions have been identified during postmarketing use of abacavir. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposures.
Body as a Whole
Redistribution/accumulation of body fat.
Cardiovascular
Myocardial infarction.
Hepatic
Lactic acidosis and hepatic steatosis [see Warnings and Precautions (5.2) ].
Skin
Suspected Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) have been reported in patients receiving abacavir primarily in combination with medications known to be associated with SJS and TEN, respectively. Because of the overlap of clinical signs and symptoms between hypersensitivity to abacavir and SJS and TEN, and the possibility of multiple drug sensitivities in some patients, abacavir should be discontinued and not restarted in such cases.
There have also been reports of erythema multiforme with abacavir use [see Adverse Reactions (6.1) ].
DRUG INTERACTIONS
Methadone
In a trial of 11 HIV-1-infected subjects receiving methadone-maintenance therapy with 600 mg of abacavir twice daily (twice the currently recommended dose), oral methadone clearance increased [see Clinical Pharmacology (12.3) ] . This alteration will not result in a methadone dose modification in the majority of patients; however, an increased methadone dose may be required in a small number of patients.
Riociguat
Coadministration with fixed-dose abacavir/dolutegravir/lamivudine resulted in increased riociguat exposure, which may increase the risk of riociguat adverse reactions [see Clinical Pharmacology (12.3) ] . The riociguat dose may need to be reduced. See full prescribing information for ADEMPAS (riociguat).
DESCRIPTION
Abacavir sulfate is a synthetic carbocyclic nucleoside analogue with inhibitory activity against HIV-1. The chemical name of abacavir sulfate is (1 S,cis)- 4-[2-amino-6-(cyclopropylamino)-9 H -purin-9-yl]-2-cyclopentene-1-methanol sulfate (salt) (2:1). Abacavir sulfate is the enantiomer with 1S , 4R absolute configuration on the cyclopentene ring. It has a molecular formula of (C 14 H 18 N 6 O) 2 •H 2 SO 4 and a molecular weight of 670.76 g per mol. It has the following structural formula:
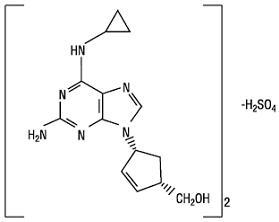
Abacavir sulfate USP is a white to off-white solid and is soluble in water.
Abacavir oral solution USP is for oral administration. Each milliliter (1 mL) of abacavir oral solution USP contains abacavir sulfate USP equivalent to 20 mg of abacavir (i.e., 20 mg per mL) as active ingredient and the following inactive ingredients: artificial strawberry and banana flavors, citric acid anhydrous, methylparaben and propylparaben (added as preservatives), propylene glycol, saccharin sodium, sodium citrate (dihydrate), noncrystallizing sorbitol solution, and water.
In vivo , abacavir sulfate dissociates to its free base, abacavir. Dosages are expressed in terms of abacavir.
CLINICAL PHARMACOLOGY
Mechanism of Action
Abacavir is an antiretroviral agent [see Microbiology (12.4) ] .
Pharmacokinetics
Pharmacokinetics in Adults
The pharmacokinetic properties of abacavir were independent of dose over the range of 300 to 1,200 mg per day. Absorption: Following oral administration, abacavir is rapidly absorbed and extensively distributed. The geometric mean absolute bioavailability of the tablet was 83%. Plasma abacavir AUC was similar following administration of the oral solution or tablets. After oral administration of 300 mg twice daily in 20 subjects, the steady-state peak serum abacavir concentration (C max ) was 3 ± 0.89 mcg per mL (mean ± SD) and AUC (0-12 h) was 6.02 ± 1.73 mcg•hour per mL. After oral administration of a single dose of 600 mg of abacavir in 20 subjects, C max was 4.26 ± 1.19 mcg per mL (mean ± SD) and AUC ∞ was 11.95 ± 2.51 mcg•hour per mL. Effect of Food: Bioavailability of abacavir tablets was assessed in the fasting and fed states with no significant difference in systemic exposure (AUC ∞ ); therefore, abacavir tablets may be administered with or without food. Systemic exposure to abacavir was comparable after administration of abacavir oral solution and abacavir tablets. Therefore, these products may be used interchangeably.
Distribution: The apparent volume of distribution after IV administration of abacavir was 0.86 ± 0.15 L per kg, suggesting that abacavir distributes into extravascular space. In 3 subjects, the CSF AUC (0-6 h) to plasma abacavir AUC (0-6 h) ratio ranged from 27% to 33%. Binding of abacavir to human plasma proteins is approximately 50% and was independent of concentration. Total blood and plasma drug-related radioactivity concentrations are identical, demonstrating that abacavir readily distributes into erythrocytes. Elimination: In single-dose trials, the observed elimination half-life (t 1/2 ) was 1.54 ± 0.63 hours. After intravenous administration, total clearance was 0.8 ± 0.24 L per hour per kg (mean ± SD). Metabolism: In humans, abacavir is not significantly metabolized by cytochrome P450 enzymes. The primary routes of elimination of abacavir are metabolism by alcohol dehydrogenase to form the 5′-carboxylic acid and glucuronyl transferase to form the 5′-glucuronide. The metabolites do not have antiviral activity. In vitro experiments reveal that abacavir does not inhibit human CYP3A4, CYP2D6, or CYP2C9 activity at clinically relevant concentrations. Excretion: Elimination of abacavir was quantified in a mass balance trial following administration of a 600 mg dose of 14 C-abacavir: 99% of the radioactivity was recovered, 1.2% was excreted in the urine as abacavir, 30% as the 5′-carboxylic acid metabolite, 36% as the 5′-glucuronide metabolite, and 15% as unidentified minor metabolites in the urine. Fecal elimination accounted for 16% of the dose. Specific Populations
Patients with Renal Impairment: The pharmacokinetic properties of abacavir have not been determined in patients with impaired renal function. Renal excretion of unchanged abacavir is a minor route of elimination in humans. Patients with Hepatic Impairment: The pharmacokinetics of abacavir have been studied in subjects with mild hepatic impairment (Child-Pugh Class A). Results showed that there was a mean increase of 89% in the abacavir AUC and an increase of 58% in the half-life of abacavir after a single dose of 600 mg of abacavir. The AUCs of the metabolites were not modified by mild liver disease; however, the rates of formation and elimination of the metabolites were decreased [see Contraindications (4), Use in Specific Populations (8.6)] . Pregnant Women: Abacavir pharmacokinetics were studied in 25 pregnant women during the last trimester of pregnancy receiving abacavir 300 mg twice daily. Abacavir exposure (AUC) during pregnancy was similar to those in postpartum and in HIV-infected non-pregnant historical controls. Consistent with passive diffusion of abacavir across the placenta, abacavir concentrations in neonatal plasma cord samples at birth were essentially equal to those in maternal plasma at delivery.
Pediatric Patients: The pharmacokinetics of abacavir have been studied after either single or repeat doses of abacavir in 169 pediatric subjects. Subjects receiving abacavir oral solution according to the recommended dosage regimen achieved plasma concentrations of abacavir similar to adults. Subjects receiving abacavir oral tablets achieved higher plasma concentrations of abacavir than subjects receiving oral solution. The pharmacokinetics of abacavir dosed once daily in HIV-1-infected pediatric subjects aged 3 months through 12 years was evaluated in 3 trials (PENTA 13 [n = 14], PENTA 15 [n = 18], and ARROW [n = 36]). All 3 trials were 2-period, crossover, open-label pharmacokinetic trials of twice- versus once-daily dosing of abacavir and lamivudine. For the oral solution as well as the tablet formulation, these 3 trials demonstrated that once-daily dosing provides comparable AUC 0-24 to twice-daily dosing of abacavir at the same total daily dose. The mean C max was approximately 1.6- to 2.3-fold higher with abacavir once-daily dosing compared with twice-daily dosing. Geriatric Patients: The pharmacokinetics of abacavir have not been studied in subjects older than 65 years. Male and Female Patients: A population pharmacokinetic analysis in HIV-1-infected male (n = 304) and female (n = 67) subjects showed no gender differences in abacavir AUC normalized for lean body weight. Racial Groups: There are no significant or clinically relevant racial differences between blacks and whites in abacavir pharmacokinetics.
Drug Interaction Studies
Effect of Abacavir on the Pharmacokinetics of Other Agents : In vitro studies have shown that abacavir has potential to inhibit CYP1A1 and limited potential to inhibit metabolism mediated by CYP3A4. Abacavir did not inhibit or induce other CYP enzymes (such as CYP2C9, or CYP2D6). Based on in vitro study results, abacavir at therapeutic drug exposures is not expected to affect the pharmacokinetics of drugs that are substrates of the following transporters: organic anion transporter polypeptide (OATP)1B1/3, breast cancer resistance protein (BCRP) or P-glycoprotein (P-gp), organic cation transporter (OCT)1, OCT2, or multidrug and toxic extrusion protein (MATE)1 and MATE2-K. Riociguat : Coadministration of a single dose of riociguat (0.5 mg) to HIV-1–infected subjects receiving fixed-dose abacavir/dolutegravir/lamivudine is reported to increase riociguat AUC (∞) compared with riociguat AUC (∞) reported in healthy subjects due to CYP1A1 inhibition by abacavir. The exact magnitude of increase in riociguat exposure has not been fully characterized based on findings from two studies [see Drug Interactions (7.2) ]. Effect of Other Agents on the Pharmacokinetics of Abacavir: In vitro , abacavir is not a substrate of OATP1B1, OAP1B3, OCT1, OCT2, OAT1, MATE1, MATE2-K, multidrug resistance-associated protein (MRP)2 or MRP4; therefore, drugs that modulate these transporters are not expected to affect abacavir plasma concentrations. Abacavir is a substrate of BCRP and P-gp in vitro ; however, considering its absolute bioavailability (83%), modulators of these transporters are unlikely to result in a clinically relevant impact on abacavir concentrations. Lamivudine and/or Zidovudine: Fifteen HIV-1-infected subjects were enrolled in a crossover-designed drug interaction trial evaluating single doses of abacavir (600 mg), lamivudine (150 mg), and zidovudine (300 mg) alone or in combination. Analysis showed no clinically relevant changes in the pharmacokinetics of abacavir with the addition of lamivudine or zidovudine or the combination of lamivudine and zidovudine. Lamivudine exposure (AUC decreased 15%) and zidovudine exposure (AUC increased 10%) did not show clinically relevant changes with concurrent abacavir. Ethanol: Abacavir has no effect on the pharmacokinetic properties of ethanol. Ethanol decreases the elimination of abacavir causing an increase in overall exposure. Due to the common metabolic pathways of abacavir and ethanol via alcohol dehydrogenase, the pharmacokinetic interaction between abacavir and ethanol was studied in 24 HIV-1-infected male subjects. Each subject received the following treatments on separate occasions: a single 600 mg dose of abacavir, 0.7 g per kg ethanol (equivalent to 5 alcoholic drinks), and abacavir 600 mg plus 0.7 g per kg ethanol. Coadministration of ethanol and abacavir resulted in a 41% increase in abacavir AUC ∞ and a 26% increase in abacavir t 1/2 . Abacavir had no effect on the pharmacokinetic properties of ethanol, so no clinically significant interaction is expected in men. This interaction has not been studied in females. Methadone: In a trial of 11 HIV-1-infected subjects receiving methadone-maintenance therapy (40 mg and 90 mg daily), with 600 mg of abacavir twice daily (twice the currently recommended dose), oral methadone clearance increased 22% (90% CI: 6% to 42%). This alteration will not result in a methadone dose modification in the majority of patients; however, an increased methadone dose may be required in a small number of patients [see Drug Interactions (7)] . The addition of methadone had no clinically significant effect on the pharmacokinetic properties of abacavir.
Microbiology
Abacavir is a carbocyclic synthetic nucleoside analogue. Abacavir is converted by cellular enzymes to the active metabolite, carbovir triphosphate (CBV-TP), an analogue of deoxyguanosine-5′-triphosphate (dGTP). CBV-TP inhibits the activity of HIV-1 reverse transcriptase (RT) both by competing with the natural substrate dGTP and by its incorporation into viral DNA.
Antiviral Activity
The antiviral activity of abacavir against HIV-1 was assessed in a number of cell lines including primary monocytes/macrophages and peripheral blood mononuclear cells (PBMCs). EC 50 values ranged from 3.7 to 5.8 microM (1 microM = 0.28 mcg per mL) and 0.07 to 1 microM against HIV-1 IIIB and HIV-1 BaL , respectively, and the mean EC 50 value was 0.26 ± 0.18 microM against 8 clinical isolates. The median EC 50 values of abacavir were 344 nM (range: 14.8 to 676 nM), 16.9 nM (range: 5.9 to 27.9 nM), 8.1 nM (range: 1.5 to 16.7 nM), 356 nM (range: 35.7 to 396 nM), 105 nM (range: 28.1 to 168 nM), 47.6 nM (range: 5.2 to 200 nM), 51.4 nM (range: 7.1 to 177 nM), and 282 nM (range: 22.4 to 598 nM) against HIV-1 clades A-G and group O viruses (n = 3 except n = 2 for clade B), respectively. The EC 50 values against HIV-2 isolates (n = 4), ranged from 0.024 to 0.49 microM. The antiviral activity of abacavir in cell culture was not antagonized when combined with the nucleoside reverse transcriptase inhibitors (NRTIs) didanosine, emtricitabine, lamivudine, stavudine, tenofovir, zalcitabine or zidovudine, the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine, or the protease inhibitor (PI) amprenavir. Ribavirin (50 microM) used in the treatment of chronic HCV infection had no effect
on the anti-HIV-1 activity of abacavir in cell culture.
Resistance
HIV-1 isolates with reduced susceptibility to abacavir have been selected in cell culture. Genotypic analysis of isolates selected in cell culture and recovered from abacavir-treated subjects demonstrated that amino acid substitutions K65R, L74V, Y115F, and M184V/I emerged in HIV-1 RT. M184V or I substitutions resulted in an approximately 2-fold decrease in susceptibility to abacavir. Substitutions K65R, L74M, or Y115F with M184V or I conferred a 7- to 8-fold reduction in abacavir susceptibility, and combinations of three substitutions were required to confer more than an 8-fold reduction in susceptibility.
Thirty-nine percent (7 of 18) of the isolates from subjects who experienced virologic failure in the abacavir once-daily arm had a greater than 2.5-fold mean decrease in abacavir susceptibility with a median-fold decrease of 1.3 (range: 0.5 to 11) compared with 29% (5 of 17) of the failure isolates in the twice-daily arm with a median-fold decrease of 0.92 (range: 0.7 to 13).
Cross-Resistance
Cross-resistance has been observed among NRTIs. Isolates containing abacavir resistance-associated substitutions, namely, K65R, L74V, Y115F, and M184V, exhibited cross-resistance to didanosine, emtricitabine, lamivudine, and tenofovir in cell culture and in subjects. An increasing number of thymidine analogue mutation substitutions (TAMs: M41L, D67N, K70R, L210W, T215Y/F, K219E/R/H/Q/N) is associated with a progressive reduction in abacavir susceptibility.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
Abacavir was administered orally at 3 dosage levels to separate groups of mice and rats in 2-year carcinogenicity studies. Results showed an increase in the incidence of malignant and non-malignant tumors. Malignant tumors occurred in the preputial gland of males and the clitoral gland of females of both species, and in the liver of female rats. In addition, non-malignant tumors also occurred in the liver and thyroid gland of female rats. These observations were made at systemic exposures in the range of 6 to 32 times the human exposure at the recommended dose of 600 mg.
Mutagenicity
Abacavir induced chromosomal aberrations both in the presence and absence of metabolic activation in an in vitro cytogenetic study in human lymphocytes. Abacavir was mutagenic in the absence of metabolic activation, although it was not mutagenic in the presence of metabolic activation in an L5178Y mouse lymphoma assay. Abacavir was clastogenic in males and not clastogenic in females in an in vivo mouse bone marrow micronucleus assay.
Abacavir was not mutagenic in bacterial mutagenicity assays in the presence and absence of metabolic activation.
Impairment of Fertility
Abacavir did not affect male or female fertility in rats at a dose associated with exposures (AUC) approximately 3.3 times (male) or 4.1 times (female) those in humans at the clinically recommended dose.
Animal Toxicology and/or Pharmacology
Myocardial degeneration was found in mice and rats following administration of abacavir for 2 years. The systemic exposures were equivalent to 7 to 24 times the expected systemic exposure in humans at a dose of 600 mg. The clinical relevance of this finding has not been determined.
CLINICAL STUDIES
Adult Trials
Therapy-Naive Adults
CNA30024 was a multicenter, double-blind, controlled trial in which 649 HIV-1-infected, therapy-naive adults were randomized and received either abacavir (300 mg twice daily), lamivudine (150 mg twice daily), and efavirenz (600 mg once daily); or zidovudine (300 mg twice daily), lamivudine (150 mg twice daily), and efavirenz (600 mg once daily). The duration of double-blind treatment was at least 48 weeks. Trial participants were male (81%), white (51%), black (21%), and Hispanic (26%). The median age was 35 years; the median pretreatment CD4+ cell count was 264 cells per mm 3 , and median plasma HIV-1 RNA was 4.79 log 10 copies per mL. The outcomes of randomized treatment are provided in Table 7.
| a Subjects achieved and maintained confirmed HIV-1 RNA less than or equal to 50 copies per mL (less than 400 copies per mL) through Week 48 (Roche AMPLICOR Ultrasensitive HIV-1 MONITOR standard test 1 PCR). b Includes viral rebound, insufficient viral response according to the investigator, and failure to achieve confirmed less than or equal to 50 copies per mL by Week 48. c Includes consent withdrawn, lost to follow up, protocol violations, those with missing data, clinical progression, and other. | ||
| Outcome | Abacavir plus Lamivudine plus Efavirenz (n = 324) | Zidovudine plus Lamivudine plus Efavirenz (n = 325) |
| Responder a Virologic failures b Discontinued due to adverse reactions Discontinued due to other reasons c | 69% (73%) 6% 14% 10% | 69% (71%) 4% 16% 11% |
After 48 weeks of therapy, the median CD4+ cell count increases from baseline were 209 cells per mm 3 in the group receiving abacavir and 155 cells per mm 3 in the zidovudine group. Through Week 48, 8 subjects (2%) in the group receiving abacavir (5 CDC classification C events and 3 deaths) and 5 subjects (2%) on the zidovudine arm (3 CDC classification C events and 2 deaths) experienced clinical disease progression. CNA3005 was a multicenter, double-blind, controlled trial in which 562 HIV-1-infected, therapy-naive adults were randomized to receive either abacavir (300 mg twice daily) plus COMBIVIR (lamivudine 150 mg/zidovudine 300 mg twice daily), or indinavir (800 mg 3 times a day) plus COMBIVIR twice daily. The trial was stratified at randomization by pre-entry plasma HIV-1 RNA 10,000 to 100,000 copies per mL and plasma HIV-1 RNA greater than 100,000 copies per mL. Trial participants were male (87%), white (73%), black (15%), and Hispanic (9%). At baseline the median age was 36 years; the median baseline CD4+ cell count was 360 cells per mm 3 , and median baseline plasma HIV-1 RNA was 4.8 log 10 copies per mL. Proportions of subjects with plasma HIV-1 RNA less than 400 copies per mL (using Roche AMPLICOR HIV-1 MONITOR Test) through 48 weeks of treatment are summarized in Table 8.
| a Subjects achieved and maintained confirmed HIV-1 RNA less than 400 copies per mL. b Includes viral rebound and failure to achieve confirmed less than 400 copies per mL by Week 48. c Includes consent withdrawn, lost to follow up, protocol violations, those with missing data, clinical progression, and other. | ||
| Outcome | Abacavir plus Lamivudine/ Zidovudine (n = 262) | Indinavir plus Lamivudine/ Zidovudine (n = 265) |
| Responder a Virologic failure b Discontinued due to adverse reactions Discontinued due to other reasons c | 49% 31% 10% 11% | 50% 28% 12% 10% |
Treatment response by plasma HIV-1 RNA strata is shown in Table 9.
| Screening HIV-1 RNA (copies/mL) | Abacavir plus Lamivudine/ Zidovudine (n = 262) | Indinavir plus Lamivudine/ Zidovudine (n = 265) | ||
| <400 copies/mL | n | <400 copies/mL | n | |
| ≥10,000 to ≤100,000 >100,000 | 50% 48% | 166 96 | 48% 52% | 165 100 |
In subjects with baseline viral load greater than 100,000 copies per mL, percentages of subjects with HIV-1 RNA levels less than 50 copies per mL were 31% in the group receiving abacavir versus 45% in the group receiving indinavir. Through Week 48, an overall mean increase in CD4+ cell count of about 150 cells per mm 3 was observed in both treatment arms. Through Week 48, 9 subjects (3.4%) in the group receiving abacavir (6 CDC classification C events and 3 deaths) and 3 subjects (1.5%) in the group receiving indinavir (2 CDC classification C events and 1 death) experienced clinical disease progression. CNA30021 was an international, multicenter, double-blind, controlled trial in which 770 HIV-1-infected, therapy-naive adults were randomized and received either abacavir 600 mg once daily or abacavir 300 mg twice daily, both in combination with lamivudine 300 mg once daily and efavirenz 600 mg once daily. The double-blind treatment duration was at least 48 weeks. Trial participants had a mean age of 37 years; were male (81%), white (54%), black (27%), and American Hispanic (15%). The median baseline CD4+ cell count was 262 cells per mm 3 (range: 21 to 918 cells per mm 3 ) and the median baseline plasma HIV-1 RNA was 4.89 log 10 copies per mL (range: 2.6 to 6.99 log 10 copies per mL).
The outcomes of randomized treatment are provided in Table 10.
| a Subjects achieved and maintained confirmed HIV-1 RNA less than 50 copies per mL (less than 400 copies per mL) through Week 48 (Roche AMPLICOR Ultrasensitive HIV-1 MONITOR standard test version 1). b Includes viral rebound, failure to achieve confirmed less than 50 copies per mL (less than 400 copies per mL) by Week 48, and insufficient viral load response. c Includes consent withdrawn, lost to follow up, protocol violations, clinical progression, and other. | ||
| Outcome | Abacavir 600 mg q.d. plus EPIVIR plus Efavirenz (n = 384) | Abacavir 300 mg b.i.d. plus EPIVIR plus Efavirenz (n = 386) |
| Responder a Virologic failure b Discontinued due to adverse reactions Discontinued due to other reasons c | 64% (71%) 11% (5%) 13% 11% | 65% (72%) 11% (5%) 11% 13% |
After 48 weeks of therapy, the median CD4+ cell count increases from baseline were 188 cells per mm 3 in the group receiving abacavir 600 mg once daily and 200 cells per mm 3 in the group receiving abacavir 300 mg twice daily. Through Week 48, 6 subjects (2%) in the group receiving abacavir 600 mg once daily (4 CDC classification C events and 2 deaths) and 10 subjects (3%) in the group receiving abacavir 300 mg twice daily (7 CDC classification C events and 3 deaths) experienced clinical disease progression. None of the deaths were attributed to trial medications.
Pediatric Trials
Therapy-Experienced Pediatric Subjects
CNA3006 was a randomized, double-blind trial comparing abacavir 8 mg per kg twice daily plus lamivudine 4 mg per kg twice daily plus zidovudine 180 mg per m 2 twice daily versus lamivudine 4 mg per kg twice daily plus zidovudine 180 mg per m 2 twice daily. Two hundred and five therapy-experienced pediatric subjects were enrolled: female (56%), white (17%), black (50%), Hispanic (30%), median age of 5.4 years, baseline CD4+ cell percent greater than 15% (median = 27%), and median baseline plasma HIV-1 RNA of 4.6 log 10 copies per mL. Eighty percent and 55% of subjects had prior therapy with zidovudine and lamivudine, respectively, most often in combination. The median duration of prior nucleoside analogue therapy was 2 years. At 16 weeks, the proportion of subjects responding based on plasma HIV-1 RNA less than or equal to 400 copies per mL was significantly higher in subjects receiving abacavir plus lamivudine plus zidovudine compared with subjects receiving lamivudine plus zidovudine, 13% versus 2%, respectively. Median plasma HIV-1 RNA changes from baseline were -0.53 log 10 copies per mL in the group receiving abacavir plus lamivudine plus zidovudine compared with -0.21 log 10 copies per mL in the group receiving lamivudine plus zidovudine. Median CD4+ cell count increases from baseline were 69 cells per mm 3 in the group receiving abacavir plus lamivudine plus zidovudine and 9 cells per mm 3 in the group receiving lamivudine plus zidovudine.
Once-Daily Dosing
ARROW (COL105677) was a 5-year randomized, multicenter trial which evaluated multiple aspects of clinical management of HIV-1 infection in pediatric subjects. HIV-1–infected, treatment-naive subjects aged 3 months to 17 years were enrolled and treated with a first-line regimen containing abacavir and lamivudine, dosed twice daily according to World Health Organization recommendations. After a minimum of 36 weeks of treatment, subjects were given the option to participate in Randomization 3 of the ARROW trial, comparing the safety and efficacy of once-daily dosing with twice-daily dosing of abacavir and lamivudine, in combination with a third antiretroviral drug, for an additional 96 weeks. Of the 1,206 original ARROW subjects, 669 participated in Randomization 3. Virologic suppression was not a requirement for participation at baseline for Randomization 3 (following a minimum of 36 weeks of twice-daily treatment), 75% of subjects in the twice-daily cohort were virologically suppressed compared with 71% of subjects in the once-daily cohort.
The proportions of subjects with HIV-1 RNA less than 80 copies per mL through 96 weeks are shown in Table 11. The differences between virologic responses in the two treatment arms were comparable across baseline characteristics for gender and age.
| a Analyses were based on the last observed viral load data within the Week 96 window. b Predicted difference (95% CI) of response rate is -4.5% (-11% to 2%) at Week 96. c Includes subjects who discontinued due to lack or loss of efficacy or for reasons other than an adverse event or death, and who had a viral load value of greater than or equal to 80 copies per mL, or subjects who had a switch in background regimen that was not permitted by the protocol. d Other includes reasons such as withdrew consent, loss to follow-up, etc. and the last available HIV-1 RNA less than 80 copies per mL (or missing). | ||
| Outcome | Abacavir plus Lamivudine Twice-Daily Dosing (n=333) | Abacavir plus Lamivudine Once-Daily Dosing (n=336) |
| HIV-1 RNA <80 copies/mL b HIV-1 RNA ≥80 copies/mL c No virologic data Discontinued due to adverse event or death Discontinued study for other reasons d Missing data during window but on study | 70% 28% 1% 0% 1% | 67% 31% <1% <1% 1% |
HOW SUPPLIED/STORAGE AND HANDLING
Abacavir Oral Solution USP: It is a clear to opalescent, yellowish, strawberry-banana-flavored liquid. Each mL of the solution contains abacavir sulfate USP equivalent to 20 mg of abacavir. It is packaged in opaque bottles with child-resistant closure. This product does not require reconstitution.
Bottles of 240 mL NDC 65862-089-24
Store at 20 ° to 25 °C (68° to 77 °F). [see USP Controlled Room Temperature ]. DO NOT FREEZE. May be refrigerated.
Mechanism of Action
Abacavir is an antiretroviral agent [see Microbiology (12.4) ] .